Journal of Human Genetics ( IF 2.6 ) Pub Date : 2020-03-16 , DOI: 10.1038/s10038-020-0739-5 Shuying Cai 1, 2 , Jinliang Li 1 , Ye Wu 1 , Yuwu Jiang 1
|
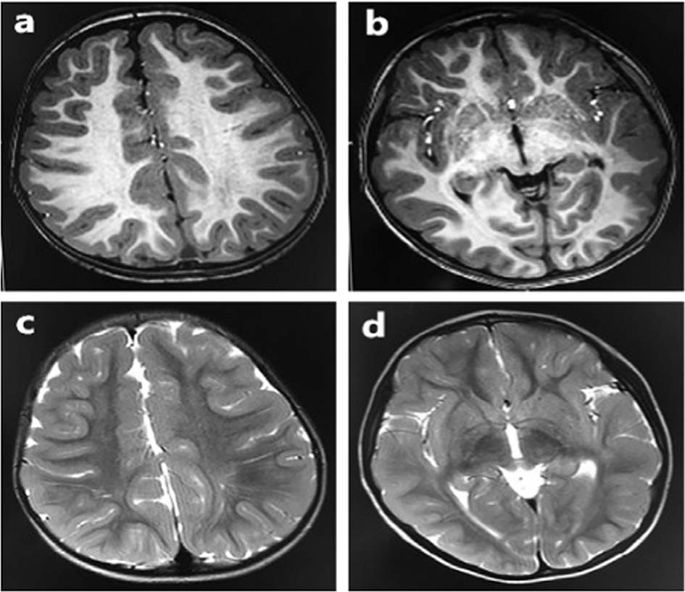
We analyzed our two new cases of infantile-onset epilepsy with developmental delay with de novo variant in TUBB2A and review the related literatures. Our two probands were both girls with infantile-onset epilepsy and global developmental delay. Case 1 had a novel de novo heterozygous missense variant: c.728C>T [p.Pro243Leu] (NM_001069.2). Her brain magnetic resonance imaging (MRI) showed nonspecific white matter myelination delay and slightly enlarged anterior horn of lateral ventricle. Her epilepsy had been controlled by TPM monotherapy. Case 2 had a reported de novo variant c.743C>T [p.Ala248Val] (NM_001069.2). Her brain MRI showed bilateral microgyria and corpus callosum dysplasia. A total of seven TUBB2A mutations cases had been published previously in five papers, therefore, until now, there were nine patients with TUBB2A mutations. All patients had developmental delay, among them seven cases also with infantile-onset epilepsy, one case with abnormal EEG but without clinical seizures. There are six cases that have different degree of cortical dysplasia, one case with cerebellar vermis atrophy and brainstem sacsinopathy, the rest two cases have no obvious brain structural abnormalities. There was one case with variant c.1249G>A (p.D417N) that had atypical clinical presentation, including prominent progressive spastic ataxia, sensory motor axonal neuropathy, and bilateral optic macular dystrophy, but relatively mild intellectual disability, his MRI showed cerebellar atrophy, thinning of the corpus callosum and pons sacsinopathy, but no cortical malformation. The p.A248V mutation was the most common mutation occurred in three patients (3/9). The clinical phenotypes of these three patients were similar, all of them had global developmental delay with no language and corpus callosum dysplasia, two cases with epilepsy and the other one only have EEG epileptic discharges without clinical seizure, two cases with cortical dysplasia and the other one without obvious brain malformation. In brief, global developmental delay was the most common phenotype of TUBB2A mutation-related disease, most cases also had infantile-onset epilepsy and cortical dysplasia and corpus callosum dysplasia. The region between seventh and eighth alpha-helix of TUBB2A may be a “hot spot” mutation domain.
中文翻译:
TUBB2A的从头突变导致婴儿发作性癫痫和发育延迟。
我们分析了我们在TUBB2A中出现的具有新变异的发育迟缓的小儿癫痫发作的两个新病例,并复习了相关文献。我们的两个先证者都是患有婴儿发作性癫痫和整体发育迟缓的女孩。情况1有一个新颖的从头杂合错义变异体:c.728C> T [p.Pro243Leu](NM_001069.2)。她的脑磁共振成像(MRI)显示非特异性白质髓鞘形成延迟,并且侧脑室前角略微增大。TPM单一疗法已控制了她的癫痫病。病例2有报道的从头变异c.743C> T [p.Ala248Val](NM_001069.2)。她的脑部MRI显示双侧小神经回和and体发育异常。共有七个TUBB2A突变案例先前已在五篇论文中发表,因此,迄今为止,已有9例TUBB2A患者突变。所有患者都有发育迟缓,其中7例还患有婴儿发作性癫痫,1例脑电图异常但无临床发作。皮质发育异常程度不同的有6例,其中小脑mis部萎缩和脑干囊性病变为1例,其余2例无明显的脑结构异常。有一例c.1249G> A(p.D417N)变体的临床表现不典型,包括突出的进行性痉挛性共济失调,感觉性运动性轴索神经病和双侧视黄斑营养不良,但智力较轻,他的MRI显示小脑萎缩,call体变薄和桥囊性病变,但没有皮质畸形。p.A248V突变是三例患者中最常见的突变(3/9)。这三例患者的临床表型相似,均具有整体发育迟缓,无语言和体发育异常,两例患有癫痫病,另一例仅伴有脑电图癫痫放电但无临床发作,两例患有皮质发育不良,另一例一无明显脑畸形。简而言之,全球发育迟缓是最常见的表型。TUBB2A突变相关疾病,大多数病例还患有婴儿发作性癫痫和皮质发育异常以及体发育异常。TUBB2A的第七个和第八个α-螺旋之间的区域可能是“热点”突变域。










































 京公网安备 11010802027423号
京公网安备 11010802027423号