当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Identifying Significant Metabolic Pathways Using Multi-Block Partial Least-Squares Analysis.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jproteome.9b00793 Lingli Deng 1 , Fanjing Guo 2 , Kian-Kai Cheng 3 , Jiangjiang Zhu 4 , Haiwei Gu 4 , Daniel Raftery 4 , Jiyang Dong 2
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jproteome.9b00793 Lingli Deng 1 , Fanjing Guo 2 , Kian-Kai Cheng 3 , Jiangjiang Zhu 4 , Haiwei Gu 4 , Daniel Raftery 4 , Jiyang Dong 2
Affiliation
|
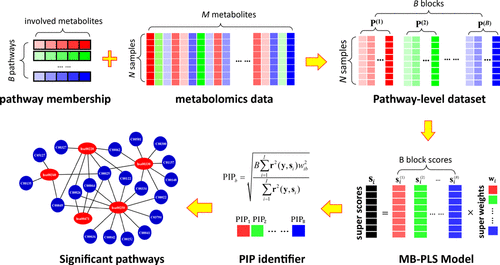
In metabolomics, identification of metabolic pathways altered by disease, genetics, or environmental perturbations is crucial to uncover the underlying biological mechanisms. A number of pathway analysis methods are currently available, which are generally based on equal-probability, topological-centrality, or model-separability methods. In brief, prior identification of significant metabolites is needed for the first two types of methods, while each pathway is modeled separately in the model-separability-based methods. In these methods, interactions between metabolic pathways are not taken into consideration. The current study aims to develop a novel metabolic pathway identification method based on multi-block partial least squares (MB-PLS) analysis by including all pathways into a global model to facilitate biological interpretation. The detected metabolites are first assigned to pathway blocks based on their roles in metabolism as defined by the KEGG pathway database. The metabolite intensity or concentration data matrix is then reconstructed as data blocks according to the metabolite subsets. Then, a MB-PLS model is built on these data blocks. A new metric, named the pathway importance in projection (PIP), is proposed for evaluation of the significance of each metabolic pathway for group separation. A simulated dataset was generated by imposing artificial perturbation on four pre-defined pathways of the healthy control group of a colorectal cancer study. Performance of the proposed method was evaluated and compared with seven other commonly used methods using both an actual metabolomics dataset and the simulated dataset. For the real metabolomics dataset, most of the significant pathways identified by the proposed method were found to be consistent with the published literature. For the simulated dataset, the significant pathways identified by the proposed method are highly consistent with the pre-defined pathways. The experimental results demonstrate that the proposed method is effective for identification of significant metabolic pathways, which may facilitate biological interpretation of metabolomics data.
中文翻译:

使用多区偏最小二乘分析识别重要的代谢途径。
在代谢组学中,识别因疾病、遗传学或环境扰动而改变的代谢途径对于揭示潜在的生物学机制至关重要。目前有多种路径分析方法,通常基于等概率、拓扑中心性或模型可分离性方法。简而言之,前两种方法需要事先识别重要的代谢物,而在基于模型可分离性的方法中,每个途径都是单独建模的。在这些方法中,没有考虑代谢途径之间的相互作用。目前的研究旨在开发一种基于多块偏最小二乘(MB-PLS)分析的新型代谢途径识别方法,将所有途径纳入全局模型以促进生物学解释。检测到的代谢物首先根据 KEGG 通路数据库定义的代谢作用分配给通路模块。然后根据代谢物子集将代谢物强度或浓度数据矩阵重建为数据块。然后,在这些数据块上建立MB-PLS模型。提出了一种新的度量,称为预测路径重要性(PIP),用于评估每个代谢路径对群体分离的重要性。通过对结直肠癌研究的健康对照组的四个预定义路径施加人工扰动来生成模拟数据集。使用实际代谢组学数据集和模拟数据集评估了所提出方法的性能,并与其他七种常用方法进行了比较。 对于真实的代谢组学数据集,发现所提出的方法确定的大多数重要途径与已发表的文献一致。对于模拟数据集,所提出的方法识别的重要路径与预定义的路径高度一致。实验结果表明,该方法可有效识别重要的代谢途径,这可能有助于代谢组学数据的生物学解释。
更新日期:2020-03-16
中文翻译:
使用多区偏最小二乘分析识别重要的代谢途径。
在代谢组学中,识别因疾病、遗传学或环境扰动而改变的代谢途径对于揭示潜在的生物学机制至关重要。目前有多种路径分析方法,通常基于等概率、拓扑中心性或模型可分离性方法。简而言之,前两种方法需要事先识别重要的代谢物,而在基于模型可分离性的方法中,每个途径都是单独建模的。在这些方法中,没有考虑代谢途径之间的相互作用。目前的研究旨在开发一种基于多块偏最小二乘(MB-PLS)分析的新型代谢途径识别方法,将所有途径纳入全局模型以促进生物学解释。检测到的代谢物首先根据 KEGG 通路数据库定义的代谢作用分配给通路模块。然后根据代谢物子集将代谢物强度或浓度数据矩阵重建为数据块。然后,在这些数据块上建立MB-PLS模型。提出了一种新的度量,称为预测路径重要性(PIP),用于评估每个代谢路径对群体分离的重要性。通过对结直肠癌研究的健康对照组的四个预定义路径施加人工扰动来生成模拟数据集。使用实际代谢组学数据集和模拟数据集评估了所提出方法的性能,并与其他七种常用方法进行了比较。 对于真实的代谢组学数据集,发现所提出的方法确定的大多数重要途径与已发表的文献一致。对于模拟数据集,所提出的方法识别的重要路径与预定义的路径高度一致。实验结果表明,该方法可有效识别重要的代谢途径,这可能有助于代谢组学数据的生物学解释。









































 京公网安备 11010802027423号
京公网安备 11010802027423号