当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Development and Evaluation of MM/GBSA Based on a Variable Dielectric GB Model for Predicting Protein-Ligand Binding Affinities.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jcim.0c00024 Ercheng Wang 1 , Hui Liu 1 , Junmei Wang 2 , Gaoqi Weng 1 , Huiyong Sun 1 , Zhe Wang 1 , Yu Kang 1 , Tingjun Hou 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jcim.0c00024 Ercheng Wang 1 , Hui Liu 1 , Junmei Wang 2 , Gaoqi Weng 1 , Huiyong Sun 1 , Zhe Wang 1 , Yu Kang 1 , Tingjun Hou 1
Affiliation
|
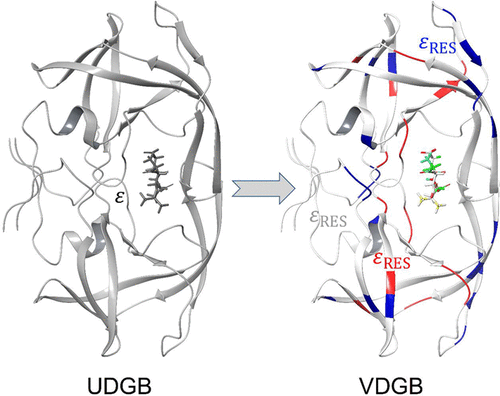
In structure-based drug design (SBDD), the molecular mechanics generalized Born surface area (MM/GBSA) approach has been widely used in ranking the binding affinity of small molecule ligands. However, an accurate estimation of protein–ligand binding affinity still remains a challenge due to the intrinsic limitation of the standard generalized Born (GB) model used in MM/GBSA. In this study, we proposed and evaluated the MM/GBSA approach based on a variable dielectric generalized Born (VDGB) model using residue-type-based dielectric constants. In the VDGB model, different dielectric values were assigned for the three types of protein residues, and the magnitude of the dielectric constants for residue types follows this order: charged ≥ polar ≥ nonpolar. We found that MM/GBSA based on a VDGB model (MM/GBSAVDGB) with an optimal dielectric constant of 4.0 for the charged residues and 1.0 for the noncharged residues together with a net-charge-dependent dielectric value for ligands achieved better predictions as judged by Pearson’s correlation coefficient than the standard MM/GBSA with a uniform solute dielectric constant of 4.0 for the training set of 130 protein–ligand complexes. The prediction on the test set with 165 protein–ligand complexes also validated the better performance of MM/GBSAVDGB. Moreover, this method exhibited potential in predicting the relative binding free energies for multiple ligands against the same target. Furthermore, we found that rational truncation of protein residues far from the binding site can significantly speed up the MM/GBSAVDGB calculations, while it almost does not influence the prediction accuracy. Therefore, it is feasible to implement the system-truncated MM/GBSAVDGB as a scoring function for SBDD.
中文翻译:

基于可变介电GB模型的MM / GBSA的开发和评估,用于预测蛋白-配体结合亲和力。
在基于结构的药物设计(SBDD)中,分子力学广义Born表面积(MM / GBSA)方法已广泛用于对小分子配体的结合亲和力进行排名。但是,由于MM / GBSA中使用的标准广义Born(GB)模型的固有局限性,准确估计蛋白质与配体的结合亲和力仍然是一个挑战。在这项研究中,我们基于残基类型的介电常数,基于可变电介质广义Born(VDGB)模型,提出并评估了MM / GBSA方法。在VDGB模型中,为三种类型的蛋白质残基分配了不同的介电值,并且残基类型的介电常数的大小遵循以下顺序:带电≥极性≥非极性。我们发现基于VDGB模型的MM / GBSA(MM / GBSA VDGB)的最佳介电常数(带电残基的最佳介电常数为4.0,不带电残基的最佳介电常数)以及配体的取决于净电荷的介电常数,通过皮尔逊相关系数判断,与具有均匀溶质介电常数的标准MM / GBSA相比,可以更好地预测130个蛋白质-配体复合物训练集的常数为4.0。对具有165种蛋白质-配体复合物的测试集的预测也验证了MM / GBSA VDGB的更好性能。而且,该方法在预测针对同一靶标的多个配体的相对结合自由能方面具有潜力。此外,我们发现远离结合位点的蛋白质残基的合理截断可以显着加快MM / GBSA VDGB计算,虽然它几乎不影响预测精度。因此,将系统截断的MM / GBSA VDGB实现为SBDD的评分功能是可行的。
更新日期:2020-03-16
中文翻译:
基于可变介电GB模型的MM / GBSA的开发和评估,用于预测蛋白-配体结合亲和力。
在基于结构的药物设计(SBDD)中,分子力学广义Born表面积(MM / GBSA)方法已广泛用于对小分子配体的结合亲和力进行排名。但是,由于MM / GBSA中使用的标准广义Born(GB)模型的固有局限性,准确估计蛋白质与配体的结合亲和力仍然是一个挑战。在这项研究中,我们基于残基类型的介电常数,基于可变电介质广义Born(VDGB)模型,提出并评估了MM / GBSA方法。在VDGB模型中,为三种类型的蛋白质残基分配了不同的介电值,并且残基类型的介电常数的大小遵循以下顺序:带电≥极性≥非极性。我们发现基于VDGB模型的MM / GBSA(MM / GBSA VDGB)的最佳介电常数(带电残基的最佳介电常数为4.0,不带电残基的最佳介电常数)以及配体的取决于净电荷的介电常数,通过皮尔逊相关系数判断,与具有均匀溶质介电常数的标准MM / GBSA相比,可以更好地预测130个蛋白质-配体复合物训练集的常数为4.0。对具有165种蛋白质-配体复合物的测试集的预测也验证了MM / GBSA VDGB的更好性能。而且,该方法在预测针对同一靶标的多个配体的相对结合自由能方面具有潜力。此外,我们发现远离结合位点的蛋白质残基的合理截断可以显着加快MM / GBSA VDGB计算,虽然它几乎不影响预测精度。因此,将系统截断的MM / GBSA VDGB实现为SBDD的评分功能是可行的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号