当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Modeling of a 14 kDa RUVBL2-Binding Domain with Medium Resolution Cryo-EM Density.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jcim.9b01095 Carlos F Rodríguez 1 , Mohinder Pal 2 , Hugo Muñoz-Hernandez 1 , Laurence H Pearl 2 , Oscar Llorca 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jcim.9b01095 Carlos F Rodríguez 1 , Mohinder Pal 2 , Hugo Muñoz-Hernandez 1 , Laurence H Pearl 2 , Oscar Llorca 1
Affiliation
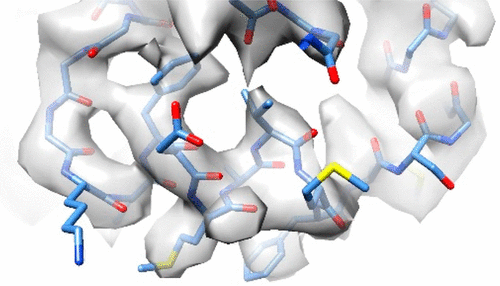
|
The number of high-resolution structures of protein complexes obtained using cryo-electron microscopy (cryo-EM) is increasing rapidly. Cryo-EM maps of large macromolecular complexes frequently contain regions resolved at different resolution levels, and modeling atomic structures de novo can be difficult for domains determined at worse than 5 Å in the absence of atomic information from other structures. Here we describe the details and step-by-step decisions in the strategy we followed to model the RUVBL2-binding domain (RBD), a 14 kDa domain at the C-terminus of RNA Polymerase II associated protein 3 (RPAP3) for which atomic information was not available. Modeling was performed on a cryo-EM map at 4.0–5.5 Å resolution, integrating information from secondary structure predictions, homology modeling, restraints from cross-linked mass spectrometry, and molecular dynamics (MD) in AMBER. Here, we compare our model with the structure of RBD determined by NMR to evaluate our strategy. We also perform new MD simulations to describe important residues mediating the interaction of RBD with RUVBL2 and analyze their conservation in RBD homologous domains. Our approach and its evaluation can serve as an example to address the analysis of medium resolution regions in cryo-EM maps.
中文翻译:

具有中等分辨率Cryo-EM密度的14 kDa RUVBL2结合域的建模。
使用冷冻电子显微镜(cryo-EM)获得的蛋白质复合物的高分辨率结构的数量正在迅速增加。大分子复合物的Cryo-EM谱图经常包含以不同分辨率级别分辨的区域,并且从头建模原子结构在没有其他结构的原子信息的情况下,对于小于5Å的畴,很难确定。在这里,我们描述了我们用来模拟RUVBL2结合域(RBD)的策略的详细信息和分步决策,该区域是位于RNA聚合酶II相关蛋白3(RPAP3)C端的14 kDa结构域,信息不可用。建模是在4.0-5.5Å分辨率的cryo-EM图上进行的,整合了二级结构预测,同源性建模,交联质谱的限制以及AMBER中的分子动力学(MD)的信息。在这里,我们将我们的模型与通过NMR确定的RBD结构进行比较,以评估我们的策略。我们还执行新的MD模拟,以描述介导RBD与RUVBL2相互作用的重要残基,并分析其在RBD同源域中的保守性。我们的方法及其评估可以作为一个实例,以解决冷冻电磁图中中等分辨率区域的分析问题。
更新日期:2020-03-16
中文翻译:
具有中等分辨率Cryo-EM密度的14 kDa RUVBL2结合域的建模。
使用冷冻电子显微镜(cryo-EM)获得的蛋白质复合物的高分辨率结构的数量正在迅速增加。大分子复合物的Cryo-EM谱图经常包含以不同分辨率级别分辨的区域,并且从头建模原子结构在没有其他结构的原子信息的情况下,对于小于5Å的畴,很难确定。在这里,我们描述了我们用来模拟RUVBL2结合域(RBD)的策略的详细信息和分步决策,该区域是位于RNA聚合酶II相关蛋白3(RPAP3)C端的14 kDa结构域,信息不可用。建模是在4.0-5.5Å分辨率的cryo-EM图上进行的,整合了二级结构预测,同源性建模,交联质谱的限制以及AMBER中的分子动力学(MD)的信息。在这里,我们将我们的模型与通过NMR确定的RBD结构进行比较,以评估我们的策略。我们还执行新的MD模拟,以描述介导RBD与RUVBL2相互作用的重要残基,并分析其在RBD同源域中的保守性。我们的方法及其评估可以作为一个实例,以解决冷冻电磁图中中等分辨率区域的分析问题。










































 京公网安备 11010802027423号
京公网安备 11010802027423号