当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
New Protocol for Predicting the Ligand-Binding Site and Mode Based on the 3D-RISM/KH Theory
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jctc.9b01069 Masatake Sugita 1, 2 , Masataka Hamano 1 , Kota Kasahara 1 , Takeshi Kikuchi 1 , Fumio Hirata 3
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jctc.9b01069 Masatake Sugita 1, 2 , Masataka Hamano 1 , Kota Kasahara 1 , Takeshi Kikuchi 1 , Fumio Hirata 3
Affiliation
|
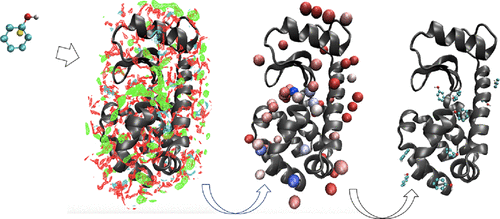
An efficient algorithm to find the binding position and mode of small ligands bound at an active site of protein is proposed based on the spatial distribution function (SDF) obtained from the three-dimensional reference interaction site model (3D-RISM) theory with the Kovalenko–Hirata (KH) closure relation. The ligand examined includes hydrophobic, acidic, and basic molecules and zwitterions. Eighteen different types of proteins, which serve as targets for those ligands, are selected to examine the robustness of the algorithm. An imaginary atom, referred to as an “anchor site”, is defined at the center of geometry of a ligand molecule that serves as a center for searching the binding position and mode of the ligand molecule in the translational and rotational spaces. The probable binding sites (PBSs) are identified based on the SDFs of the ligand molecules around the protein, and the PBS is ranked according to the peak height of SDF. The deviations from the mean height of the peak values of SDFs for 50 PBSs are analyzed based on the z-score, which is a measure of prominence of the site. The PBS found at the closest distance from the anchor site of the crystal structure is referred to as the “nearest site”. The orientation of the ligand molecule at each PBS is explored by changing the Euler angles, and the most probable binding mode is determined based on the superposition approximation. The binding position of ligand molecules is successfully predicted as one of the distinct peaks in SDF of the anchor site, with a few exceptions. The binding mode of the ligand molecule predicted based on the superposition approximation is consistent with the X-ray crystal structure in nine systems, a half of the systems investigated. The significance of the results is discussed in detail. An application of the new protocol to fragment-based drug discovery is suggested.
中文翻译:

基于3D-RISM / KH理论的配体结合位点和模式预测新协议
基于从三维参考相互作用位点模型(3D-RISM)理论与科瓦连科获得的空间分布函数(SDF),提出了一种有效的算法来寻找结合在蛋白质活性位点上的小配体的结合位置和模式–平田(KH)闭合关系。检查的配体包括疏水,酸性和碱性分子和两性离子。选择了18种不同类型的蛋白质作为这些配体的靶标,以检查算法的鲁棒性。假想原子,称为“锚定位点”,定义在配体分子几何中心,该中心用作搜索配体分子在平移和旋转空间中的结合位置和模式的中心。根据蛋白质周围配体分子的SDF识别可能的结合位点(PBS),并根据SDF的峰高对PBS进行排名。根据50个PBS分析SDF峰值的平均高度偏差。ž-score,这是衡量网站知名度的指标。在距晶体结构的锚定位置最近的距离处发现的PBS被称为“最近的位置”。通过改变欧拉角来探索每个PBS上配体分子的方向,并根据叠加近似值确定最可能的结合方式。除了少数例外,配体分子的结合位置被成功预测为锚定位点SDF中的不同峰之一。基于叠加近似预测的配体分子的结合模式与九个系统(研究的系统的一半)中的X射线晶体结构一致。详细讨论了结果的意义。建议将新协议应用于基于片段的药物发现。
更新日期:2020-04-24
中文翻译:
基于3D-RISM / KH理论的配体结合位点和模式预测新协议
基于从三维参考相互作用位点模型(3D-RISM)理论与科瓦连科获得的空间分布函数(SDF),提出了一种有效的算法来寻找结合在蛋白质活性位点上的小配体的结合位置和模式–平田(KH)闭合关系。检查的配体包括疏水,酸性和碱性分子和两性离子。选择了18种不同类型的蛋白质作为这些配体的靶标,以检查算法的鲁棒性。假想原子,称为“锚定位点”,定义在配体分子几何中心,该中心用作搜索配体分子在平移和旋转空间中的结合位置和模式的中心。根据蛋白质周围配体分子的SDF识别可能的结合位点(PBS),并根据SDF的峰高对PBS进行排名。根据50个PBS分析SDF峰值的平均高度偏差。ž-score,这是衡量网站知名度的指标。在距晶体结构的锚定位置最近的距离处发现的PBS被称为“最近的位置”。通过改变欧拉角来探索每个PBS上配体分子的方向,并根据叠加近似值确定最可能的结合方式。除了少数例外,配体分子的结合位置被成功预测为锚定位点SDF中的不同峰之一。基于叠加近似预测的配体分子的结合模式与九个系统(研究的系统的一半)中的X射线晶体结构一致。详细讨论了结果的意义。建议将新协议应用于基于片段的药物发现。










































 京公网安备 11010802027423号
京公网安备 11010802027423号