当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Capturing Deuteration Effects in a Molecular Mechanics Force Field: Deuterated THF and the THF–Water Miscibility Gap
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jctc.9b01138 Rupesh Agarwal 1, 2 , Micholas Dean Smith 1, 3 , Jeremy C. Smith 1, 3
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jctc.9b01138 Rupesh Agarwal 1, 2 , Micholas Dean Smith 1, 3 , Jeremy C. Smith 1, 3
Affiliation
|
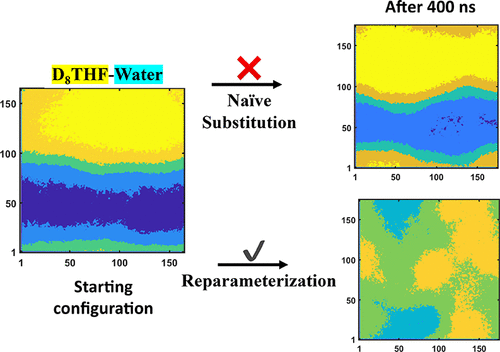
Deuteration is a common chemical modification used in conjunction with experiments such as neutron scattering, NMR, and Fourier-transform infrared for the study of molecular systems. Under the Born–Oppenheimer (BO) approximation, while the underlying potential energy surface remains unchanged by isotopic substitutions, isotopic substitution still alters intramolecular vibrations, which in turn may alter intermolecular interactions. Molecular mechanics (MM) force fields used in classical molecular dynamics simulations are assumed to represent local approximations of the BO potential energy surfaces, and hence, MD simulations using simple isotopic mass substitutions should capture BO-compatible isotope effects. However, standard MM force-field parameterizations do not directly fit to the local harmonic quantum mechanical (QM) Hessian that describes the BO surface, but rather to QM normal-modes and/or mass-dependent internal-coordinate derived distortion energies. Here, using tetrahydrofuran (THF)–water mixtures as our model system, we show that not only does a simple mass-substitution approach fail to capture an experimentally characterized deuteration effect (the loss of the closed-loop miscibility gap associated with the complete deuteration of THF) but also it is necessary to generate new MM force-field parameters that correctly describe isotopic dependent vibrations to capture the experimental deuteration effect. We show that the origin of this failure is a result of using mass-dependent features to fit the THF MM force field, which unintentionally biases the bonded terms of the force field to represent only the isotopologue used during the original force-field parameterization. In addition, we make use of our isotopologue-corrected force field for D8THF to examine the molecular origins of the isotope-dependent loss of the THF–water miscibility gap.
中文翻译:

在分子力学力场中捕获氘代效应:氘代THF和THF与水的混溶性缺口
氘化是一种常见的化学修饰形式,与诸如中子散射,NMR和傅立叶变换红外等实验一起用于分子系统的研究。在Born–Oppenheimer(BO)近似下,尽管潜在的势能表面通过同位素取代保持不变,但同位素取代仍会改变分子内的振动,进而可能改变分子间的相互作用。假定经典分子动力学模拟中使用的分子力学(MM)力场表示BO势能面的局部近似值,因此,使用简单同位素质量替代的MD模拟应捕获与BO相容的同位素效应。然而,标准的MM力场参数化不直接适合描述BO表面的局部谐波量子力学(QM)粗麻布,而是适合QM正常模式和/或质量相关的内部坐标派生的变形能量。在这里,使用四氢呋喃(THF)-水混合物作为我们的模型系统,我们证明,不仅简单的质量置换方法无法捕获实验上表征的氘代效应(与完全氘代相关的闭环混溶间隙的损失) ,但也有必要生成新的MM力场参数,以正确描述同位素相关的振动以捕获实验氘效应。我们表明,这种失败的根源是使用质量相关特征来拟合THF MM力场的结果,它会无意中偏向力场的键合项,以仅表示原始力场参数化过程中使用的同位素。此外,我们利用D同位素同位素校正的力场8 THF用于检查THF与水混溶性间隙的同位素依赖性损失的分子起源。
更新日期:2020-04-24
中文翻译:
在分子力学力场中捕获氘代效应:氘代THF和THF与水的混溶性缺口
氘化是一种常见的化学修饰形式,与诸如中子散射,NMR和傅立叶变换红外等实验一起用于分子系统的研究。在Born–Oppenheimer(BO)近似下,尽管潜在的势能表面通过同位素取代保持不变,但同位素取代仍会改变分子内的振动,进而可能改变分子间的相互作用。假定经典分子动力学模拟中使用的分子力学(MM)力场表示BO势能面的局部近似值,因此,使用简单同位素质量替代的MD模拟应捕获与BO相容的同位素效应。然而,标准的MM力场参数化不直接适合描述BO表面的局部谐波量子力学(QM)粗麻布,而是适合QM正常模式和/或质量相关的内部坐标派生的变形能量。在这里,使用四氢呋喃(THF)-水混合物作为我们的模型系统,我们证明,不仅简单的质量置换方法无法捕获实验上表征的氘代效应(与完全氘代相关的闭环混溶间隙的损失) ,但也有必要生成新的MM力场参数,以正确描述同位素相关的振动以捕获实验氘效应。我们表明,这种失败的根源是使用质量相关特征来拟合THF MM力场的结果,它会无意中偏向力场的键合项,以仅表示原始力场参数化过程中使用的同位素。此外,我们利用D同位素同位素校正的力场8 THF用于检查THF与水混溶性间隙的同位素依赖性损失的分子起源。










































 京公网安备 11010802027423号
京公网安备 11010802027423号