当前位置:
X-MOL 学术
›
J. Mater. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Template effects in Cu(I)–Bi(III) iodide double perovskites: a study of crystal structure, film orientation, band gap and photocurrent response
Journal of Materials Chemistry A ( IF 10.7 ) Pub Date : 2020/03/14 , DOI: 10.1039/d0ta00949k Le-Yu Bi 1, 2, 3, 4, 5 , Tian-Li Hu 1, 2, 3, 4, 5 , Mu-Qing Li 1, 2, 3, 4, 5 , Bo-Kai Ling 1, 2, 3, 4, 5 , Mohamed Saber Lassoued 1, 2, 3, 4, 5 , Yue-Qiao Hu 6, 7, 8, 9 , Zhaoxin Wu 10, 11, 12, 13, 14 , Guijiang Zhou 14, 15, 16, 17, 18 , Yan-Zhen Zheng 1, 2, 3, 4, 5
Journal of Materials Chemistry A ( IF 10.7 ) Pub Date : 2020/03/14 , DOI: 10.1039/d0ta00949k Le-Yu Bi 1, 2, 3, 4, 5 , Tian-Li Hu 1, 2, 3, 4, 5 , Mu-Qing Li 1, 2, 3, 4, 5 , Bo-Kai Ling 1, 2, 3, 4, 5 , Mohamed Saber Lassoued 1, 2, 3, 4, 5 , Yue-Qiao Hu 6, 7, 8, 9 , Zhaoxin Wu 10, 11, 12, 13, 14 , Guijiang Zhou 14, 15, 16, 17, 18 , Yan-Zhen Zheng 1, 2, 3, 4, 5
Affiliation
|
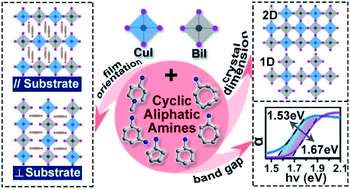
The toxicity of lead has inspired recent interest in making lead-free photovoltaic materials, among which the bimetallic double perovskite (DP) strategy is very promising. Unlike the popular Ag(I)–Bi(III) based DPs with wider band gaps ∼2.0 eV, in our previous study we found that the Cu(I)–Bi(III) method may have narrower band gaps ∼1.6 eV. Here we expand this strategy to various amines, from which we can see that the cyclic aliphatic amines (CAAs) are critical to govern the structural dimensions, thin film orientations and band gaps of the Cu(I)–Bi(III) iodide DPs. Among these compounds, CAAs with a “Z-shape” and six-member-rings result in two-dimensional structures (compounds 1–5), while CAAs with a “C-shape” and five-member-rings result in 1D bimetallic chains (compounds 6 and 7). Regardless of the structural dimensions, all seven compounds show narrow band gaps between 1.53 and 1.67 eV, which can be correlated to the distortions of the Cu–I–Bi angles. The band gap will be narrower if the Cu–I–Bi angle is closer to 180° or the octahedral distortion is smaller. Moreover, DFT calculations reveal that such band gaps are flat and direct in nature. Interestingly, the thin films formed by these compounds show preferential orientations to the ITO substrate with respect to the symmetries of the amines. For mirror-symmetric diamines the inorganic layers tend to be perpendicular to ITO; for asymmetric diamines the inorganic layers tend to be randomly oriented on ITO; for the asymmetric mono-amines the inorganic layers tend to be parallel to ITO. Such a phenomenon is, as far as we know, unknown to the field of perovskite thin film construction. To further reveal the potential of these materials for photovoltaic application we took compound 3 as a representative to study the photoelectric properties. From 20 °C to 130 °C, the conductivity of compound 3 increases over five orders of magnitude, ramping from 3.97 × 10−10 S cm−1 to 2.78 × 10−5 S cm−1, indicating a formal semiconducting behaviour. Thus, the photo-current response experiments show a ca. 17.5 nA difference between Ilight and Idark for 3, indicating the potential application of this material for light harvesting.
中文翻译:

Cu(I)–Bi(III)碘化双钙钛矿中的模板效应:晶体结构,膜取向,带隙和光电流响应的研究
铅的毒性激发了近来对制造无铅光伏材料的兴趣,其中双金属双钙钛矿(DP)策略非常有前途。与流行的基于Ag(I)–Bi(III)的DP的带隙约为2.0 eV,在我们之前的研究中,我们发现Cu(I)–Bi(III)方法的带隙可能较窄,约为1.6 eV。在这里,我们将这一策略扩展到各种胺,从中我们可以看到,环状脂肪胺(CAAs)对于控制碘化铜(I)-铋(III)DP的结构尺寸,薄膜取向和带隙至关重要。在这些化合物中,具有“ Z形”和六元环的CAA导致二维结构(化合物1-5),而带有“ C形”和五元环的CAA会生成一维双金属链(化合物6和7)。无论结构尺寸如何,所有七个化合物均显示出1.53至1.67 eV之间的窄带隙,这可能与Cu–I–Bi角的变形有关。如果Cu–I–Bi角度更接近180°或八面体变形较小,则带隙将变窄。此外,DFT计算表明,这种带隙实际上是平坦且直接的。有趣的是,相对于胺的对称性,由这些化合物形成的薄膜相对于ITO衬底显示出优先的取向。对于镜面对称的二胺,无机层倾向于垂直于ITO。对于不对称二胺,无机层倾向于在ITO上随机取向;对于不对称单胺,无机层往往与ITO平行。据我们所知,这种现象是 钙钛矿薄膜构造领域未知。为了进一步揭示这些材料在光伏应用中的潜力,我们采用了复合材料以3为代表来研究光电性能。从20°C到130°C,化合物3的电导率在五个数量级上增加,从3.97×10 -10 S cm -1逐渐增加到2.78×10 -5 S cm -1,表明这是一种正式的半导体行为。因此,光电流响应实验显示约。I亮和I暗之间的差异为17.5 nA (3倍),表明该材料可用于光收集。
更新日期:2020-04-15
中文翻译:
Cu(I)–Bi(III)碘化双钙钛矿中的模板效应:晶体结构,膜取向,带隙和光电流响应的研究
铅的毒性激发了近来对制造无铅光伏材料的兴趣,其中双金属双钙钛矿(DP)策略非常有前途。与流行的基于Ag(I)–Bi(III)的DP的带隙约为2.0 eV,在我们之前的研究中,我们发现Cu(I)–Bi(III)方法的带隙可能较窄,约为1.6 eV。在这里,我们将这一策略扩展到各种胺,从中我们可以看到,环状脂肪胺(CAAs)对于控制碘化铜(I)-铋(III)DP的结构尺寸,薄膜取向和带隙至关重要。在这些化合物中,具有“ Z形”和六元环的CAA导致二维结构(化合物1-5),而带有“ C形”和五元环的CAA会生成一维双金属链(化合物6和7)。无论结构尺寸如何,所有七个化合物均显示出1.53至1.67 eV之间的窄带隙,这可能与Cu–I–Bi角的变形有关。如果Cu–I–Bi角度更接近180°或八面体变形较小,则带隙将变窄。此外,DFT计算表明,这种带隙实际上是平坦且直接的。有趣的是,相对于胺的对称性,由这些化合物形成的薄膜相对于ITO衬底显示出优先的取向。对于镜面对称的二胺,无机层倾向于垂直于ITO。对于不对称二胺,无机层倾向于在ITO上随机取向;对于不对称单胺,无机层往往与ITO平行。据我们所知,这种现象是 钙钛矿薄膜构造领域未知。为了进一步揭示这些材料在光伏应用中的潜力,我们采用了复合材料以3为代表来研究光电性能。从20°C到130°C,化合物3的电导率在五个数量级上增加,从3.97×10 -10 S cm -1逐渐增加到2.78×10 -5 S cm -1,表明这是一种正式的半导体行为。因此,光电流响应实验显示约。I亮和I暗之间的差异为17.5 nA (3倍),表明该材料可用于光收集。










































 京公网安备 11010802027423号
京公网安备 11010802027423号