当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First principles studies of oxygen adsorption on the γ-U (1 1 0) surface and influences of Mo doping
Computational Materials Science ( IF 3.1 ) Pub Date : 2020-06-01 , DOI: 10.1016/j.commatsci.2020.109633 Xiaofeng Tian , Yu Wang , Lingshan Li , Mingde Wu , You Yu
Computational Materials Science ( IF 3.1 ) Pub Date : 2020-06-01 , DOI: 10.1016/j.commatsci.2020.109633 Xiaofeng Tian , Yu Wang , Lingshan Li , Mingde Wu , You Yu
|
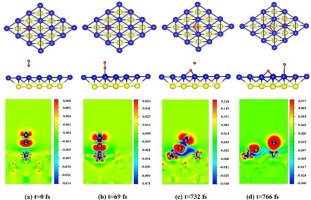
Abstract By ab initio molecular dynamics (AIMD) simulations and density-functional theory (DFT) calculations, we studied the kinetics and energetics of oxygen absorption on clean and Mo-doped γ-U (1 1 0) surface. The AIMD simulations shows that O2 will dissociates into separate O atoms, which are finally adsorbed on the surface. The doping Mo can change the final adsorption sites of O atoms. In addition, we found that temperature can accelerate O2 dissociation on γ-U (1 1 0) surface. The calculation of adsorption energies shows that the most preferred site for O atoms on the γ-U (1 1 0) surface is the short bridge site, followed by the 3-fold hollow. The doping Mo will decrease the adsorption energies of all the adsorption sites. With Mo-doping, the most preferred site for O atom is still short bridge. But, the second preferred adsorption site become long bridge. For clean and Mo-doped surfaces, the on-top is the least preferred adsorption site for atomic oxygen. Through calculations of diffusion pathways, we found Mo-doping raise the energy barrier for O atom diffusion on the surface. By the calculated segregation energies, it is found that the Mo atom prefers to locate in γ-U “bulk” rather than the top layer of the surface.
中文翻译:

γ-U(1 1 0)表面氧吸附的第一性原理研究及Mo掺杂的影响
摘要 通过从头分子动力学(AIMD) 模拟和密度泛函理论(DFT) 计算,我们研究了清洁和Mo 掺杂γ-U (1 1 0) 表面吸氧的动力学和能量学。AIMD 模拟表明,O2 将分解成单独的 O 原子,最终吸附在表面上。掺杂Mo可以改变O原子的最终吸附位点。此外,我们发现温度可以加速 γ-U (1 1 0) 表面的 O2 解离。吸附能的计算表明,γ-U (1 1 0) 表面上 O 原子最优选的位置是短桥位,其次是 3 倍空心。掺杂钼会降低所有吸附位点的吸附能。对于 Mo 掺杂,O 原子最优选的位置仍然是短桥。但,第二个优选的吸附位点成为长桥。对于清洁和 Mo 掺杂的表面,顶部是原子氧最不优选的吸附位点。通过扩散路径的计算,我们发现Mo掺杂提高了O原子在表面扩散的能垒。通过计算的偏析能,发现 Mo 原子更喜欢位于 γ-U“主体”而不是表面的顶层。
更新日期:2020-06-01
中文翻译:
γ-U(1 1 0)表面氧吸附的第一性原理研究及Mo掺杂的影响
摘要 通过从头分子动力学(AIMD) 模拟和密度泛函理论(DFT) 计算,我们研究了清洁和Mo 掺杂γ-U (1 1 0) 表面吸氧的动力学和能量学。AIMD 模拟表明,O2 将分解成单独的 O 原子,最终吸附在表面上。掺杂Mo可以改变O原子的最终吸附位点。此外,我们发现温度可以加速 γ-U (1 1 0) 表面的 O2 解离。吸附能的计算表明,γ-U (1 1 0) 表面上 O 原子最优选的位置是短桥位,其次是 3 倍空心。掺杂钼会降低所有吸附位点的吸附能。对于 Mo 掺杂,O 原子最优选的位置仍然是短桥。但,第二个优选的吸附位点成为长桥。对于清洁和 Mo 掺杂的表面,顶部是原子氧最不优选的吸附位点。通过扩散路径的计算,我们发现Mo掺杂提高了O原子在表面扩散的能垒。通过计算的偏析能,发现 Mo 原子更喜欢位于 γ-U“主体”而不是表面的顶层。










































 京公网安备 11010802027423号
京公网安备 11010802027423号