当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
TD-DFT benchmark for UV-visible spectra of fused-ring electron acceptors using global and range-separated hybrids
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020/03/11 , DOI: 10.1039/d0cp00060d Amjad Ali 1, 2, 3, 4 , Muhammad Imran Rafiq 1, 2, 3, 4 , Zhuohan Zhang 1, 2, 3, 4 , Jinru Cao 1, 2, 3, 4 , Renyong Geng 1, 2, 3, 4 , Baojing Zhou 1, 2, 3, 4 , Weihua Tang 1, 2, 3, 4
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020/03/11 , DOI: 10.1039/d0cp00060d Amjad Ali 1, 2, 3, 4 , Muhammad Imran Rafiq 1, 2, 3, 4 , Zhuohan Zhang 1, 2, 3, 4 , Jinru Cao 1, 2, 3, 4 , Renyong Geng 1, 2, 3, 4 , Baojing Zhou 1, 2, 3, 4 , Weihua Tang 1, 2, 3, 4
Affiliation
|
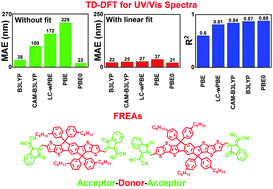
Non-fullerene acceptors, especially acceptor–donor–acceptor structured fused-ring electron acceptors (FREAs), have attracted widespread attention in organic solar cells because of their versatile molecular design in fine-tuning light absorption and energy levels. We report the accuracy of Time-Dependent Density Functional Theory (TD-DFT) for FREAs by comparing their theoretically predicted vertical absorption wavelength (λver-abso) with the experimental maximum absorption (λmax). The λver-abso values of 50 molecules obtained from major types of FREAs have been investigated using TD-DFT by considering the solvent effects. The values of λver-abso predicted with a pure density functional (PBE), global hybrids (B3LYP and PBE0) and range-separated schemes (CAM-B3LYP and LC-ωPBE) follow the exact exchange percentage included at an intermediate inter-electronic distance. Global hybrids outperform all other schemes. The mean absolute error provided is 22 nm by PBE0 and 38 nm by B3LYP for the whole set of molecules. The maximum deviation of 92 nm provided by B3LYP and 69 nm provided by PBE0 confirms that PBE0 is more appropriate for predicting the absorption wavelengths when designing new FREAs. By applying linear regression analysis to obtain the calibration curve, we found that the range-separated methods provide an equal or even more consistent description of FREA excited states. For the whole set of molecules, linearly corrected data yield an average error of 25 and 27 nm for CAM-B3LYP and LC-ωPBE, respectively. Consequently, when a statistical analysis technique is applicable for a certain series of FREAs, a theoretical method permits a chemically comprehensive and empirically good explanation of UV/Vis spectra for newly-designed FREAs.
中文翻译:

TD-DFT基准,用于使用整体和距离分隔的杂化体的稠环电子受体的紫外可见光谱
非富勒烯受体,尤其是受体-供体-受体结构的稠环电子受体(FREAs),由于其在微调光吸收和能级方面的多功能分子设计,已引起了有机太阳能电池的广泛关注。我们通过他们的理论预测垂直吸收波长(比较汇报FREAs时密度泛函理论(TD-DFT)的精度λ VER-ABSO)与实验最大吸收(λ最大)。所述λ VER-ABSO从主要类型FREAs已获得50个分子的值使用TD-DFT通过考虑溶剂效应的影响。的值λ VER-ABSO用纯密度泛函(PBE)进行预测,全局混合体(B3LYP和PBE0)和范围分离方案(CAM-B3LYP和LC-ωPBE)遵循在中间电子间距处包含的确切交换百分比。全局混合动力胜过其他所有方案。对于整个分子组,PBE0提供的平均绝对误差为22 nm,B3LYP提供的平均绝对误差为38 nm。B3LYP提供的92 nm和PBE0提供的69 nm的最大偏差证实,在设计新的FREA时,PBE0更适合于预测吸收波长。通过应用线性回归分析获得校准曲线,我们发现范围分离方法提供了对FREA激发态的相等或更一致的描述。对于整个分子 线性校正数据得出的CAM-B3LYP和LC-ωPBE的平均误差分别为25和27 nm。因此,当统计分析技术适用于某些系列的FREA时,一种理论方法可以对新设计的FREA进行化学上全面的和经验上的UV / Vis光谱解释。
更新日期:2020-04-15
中文翻译:
TD-DFT基准,用于使用整体和距离分隔的杂化体的稠环电子受体的紫外可见光谱
非富勒烯受体,尤其是受体-供体-受体结构的稠环电子受体(FREAs),由于其在微调光吸收和能级方面的多功能分子设计,已引起了有机太阳能电池的广泛关注。我们通过他们的理论预测垂直吸收波长(比较汇报FREAs时密度泛函理论(TD-DFT)的精度λ VER-ABSO)与实验最大吸收(λ最大)。所述λ VER-ABSO从主要类型FREAs已获得50个分子的值使用TD-DFT通过考虑溶剂效应的影响。的值λ VER-ABSO用纯密度泛函(PBE)进行预测,全局混合体(B3LYP和PBE0)和范围分离方案(CAM-B3LYP和LC-ωPBE)遵循在中间电子间距处包含的确切交换百分比。全局混合动力胜过其他所有方案。对于整个分子组,PBE0提供的平均绝对误差为22 nm,B3LYP提供的平均绝对误差为38 nm。B3LYP提供的92 nm和PBE0提供的69 nm的最大偏差证实,在设计新的FREA时,PBE0更适合于预测吸收波长。通过应用线性回归分析获得校准曲线,我们发现范围分离方法提供了对FREA激发态的相等或更一致的描述。对于整个分子 线性校正数据得出的CAM-B3LYP和LC-ωPBE的平均误差分别为25和27 nm。因此,当统计分析技术适用于某些系列的FREA时,一种理论方法可以对新设计的FREA进行化学上全面的和经验上的UV / Vis光谱解释。










































 京公网安备 11010802027423号
京公网安备 11010802027423号