当前位置:
X-MOL 学术
›
Mol. Omics
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Choosing proper normalization is essential for discovery of sparse glycan biomarkers.
Molecular Omics ( IF 3.0 ) Pub Date : 2020-03-10 , DOI: 10.1039/c9mo00174c Hae-Won Uh 1 , Lucija Klarić 2 , Ivo Ugrina 3 , Gordan Lauc 4 , Age K Smilde 5 , Jeanine J Houwing-Duistermaat 6
Molecular Omics ( IF 3.0 ) Pub Date : 2020-03-10 , DOI: 10.1039/c9mo00174c Hae-Won Uh 1 , Lucija Klarić 2 , Ivo Ugrina 3 , Gordan Lauc 4 , Age K Smilde 5 , Jeanine J Houwing-Duistermaat 6
Affiliation
|
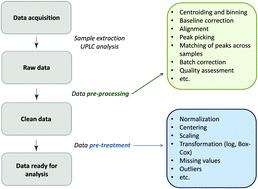
Rapid progress in high-throughput glycomics analysis enables the researchers to conduct large sample studies. Typically, the between-subject differences in total abundance of raw glycomics data are very large, and it is necessary to reduce the differences, making measurements comparable across samples. Essentially there are two ways to approach this issue: row-wise and column-wise normalization. In glycomics, the differences per subject are usually forced to be exactly zero, by scaling each sample having the sum of all glycan intensities equal to 100%. This total area (row-wise) normalization (TA) results in so-called compositional data, rendering many standard multivariate statistical methods inappropriate or inapplicable. Ignoring the compositional nature of the data, moreover, may lead to spurious results. Alternatively, a log-transformation to the raw data can be performed prior to column-wise normalization and implementing standard statistical tools. Until now, there is no clear consensus on the appropriate normalization method applied to glycomics data. Nor is systematic investigation of impact of TA on downstream analysis available to justify the choice of TA. Our motivation lies in efficient variable selection to identify glycan biomarkers with regard to accurate prediction as well as interpretability of the model chosen. Via extensive simulations we investigate how different normalization methods affect the performance of variable selection, and compare their performance. We also address the effect of various types of measurement error in glycans: additive, multiplicative and two-component error. We show that when sample-wise differences are not large row-wise normalization (like TA) can have deleterious effects on variable selection and prediction.
中文翻译:

选择适当的归一化对于发现稀疏聚糖生物标记至关重要。
高通量糖组学分析的快速进展使研究人员能够进行大量样品研究。通常,原始糖组学数据的总丰度在受试者之间的差异非常大,因此有必要减小差异,从而使各个样品的测量结果具有可比性。从本质上讲,有两种方法可以解决此问题:行标准化和列标准化。在糖组学中,通常通过缩放每个样本的所有聚糖强度之和等于100%,迫使每个受试者的差异恰好为零。该总面积(行)归一化(TA)导致了所谓的成分数据,使许多标准的多元统计方法不适当或不适用。此外,忽略数据的构成性质可能会导致虚假结果。或者,在按列规范化和实施标准统计工具之前,可以执行对原始数据的对数转换。到目前为止,对于适用于糖组学数据的适当归一化方法尚无明确共识。也没有提供关于TA对下游分析影响的系统研究来证明TA选择的合理性。我们的动机在于有效的变量选择,以根据准确的预测以及所选模型的可解释性来识别聚糖生物标志物。通过广泛的模拟,我们研究了不同的归一化方法如何影响变量选择的性能,并比较了它们的性能。我们还解决了聚糖中各种类型的测量误差的影响:加性误差,乘性误差和两组分误差。我们表明,当样本差异不大时,行归一化(如TA)会对变量选择和预测产生有害影响。
更新日期:2020-03-10
中文翻译:
选择适当的归一化对于发现稀疏聚糖生物标记至关重要。
高通量糖组学分析的快速进展使研究人员能够进行大量样品研究。通常,原始糖组学数据的总丰度在受试者之间的差异非常大,因此有必要减小差异,从而使各个样品的测量结果具有可比性。从本质上讲,有两种方法可以解决此问题:行标准化和列标准化。在糖组学中,通常通过缩放每个样本的所有聚糖强度之和等于100%,迫使每个受试者的差异恰好为零。该总面积(行)归一化(TA)导致了所谓的成分数据,使许多标准的多元统计方法不适当或不适用。此外,忽略数据的构成性质可能会导致虚假结果。或者,在按列规范化和实施标准统计工具之前,可以执行对原始数据的对数转换。到目前为止,对于适用于糖组学数据的适当归一化方法尚无明确共识。也没有提供关于TA对下游分析影响的系统研究来证明TA选择的合理性。我们的动机在于有效的变量选择,以根据准确的预测以及所选模型的可解释性来识别聚糖生物标志物。通过广泛的模拟,我们研究了不同的归一化方法如何影响变量选择的性能,并比较了它们的性能。我们还解决了聚糖中各种类型的测量误差的影响:加性误差,乘性误差和两组分误差。我们表明,当样本差异不大时,行归一化(如TA)会对变量选择和预测产生有害影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号