当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate Quantum Chemical Calculation of Ionization Potentials: Validation of the DFT-LOC Approach via a Large Data Set Obtained from Experiments and Benchmark Quantum Chemical Calculations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-09 , DOI: 10.1021/acs.jctc.9b00875 Guangqi Li 1 , Benjamin Rudshteyn 1 , James Shee 1 , John L. Weber 1 , Dilek Coskun 1 , Art D. Bochevarov 2 , Richard A. Friesner 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-09 , DOI: 10.1021/acs.jctc.9b00875 Guangqi Li 1 , Benjamin Rudshteyn 1 , James Shee 1 , John L. Weber 1 , Dilek Coskun 1 , Art D. Bochevarov 2 , Richard A. Friesner 1
Affiliation
|
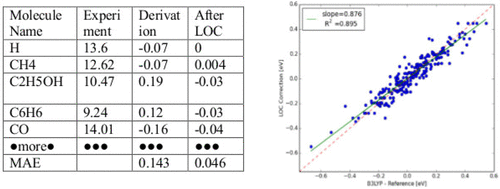
Density functional theory (DFT) is known to often fail when calculating thermodynamic values, such as ionization potentials (IPs), due to nondynamical error (i.e., the self-interaction term). Localized orbital corrections (LOCs), derived from assigning corresponding corrections for the atomic orbitals, bonds, and paired and unpaired electrons, are utilized to correct the IPs calculated from DFT. Some of the assigned parameters, which are physically due to the contraction of and change of the environment around a bond, depend on identifying the location in the molecule from which the electron is removed using differences in the charge density between neutral and oxidized species. In our training set, various small organic and inorganic molecules from the literature with the reported experimental IP were collected using the NIST database. For certain molecules with uncertain or no experimental measurements, we obtain the IP using coupled cluster theory and auxiliary field quantum Monte Carlo. After applying these corrections, as generated by least-squares regression, LOC reduces the mean absolute deviation (MAD) of the training set from 0.143 to 0.046 eV (R2 = 0.895), and LOC reduces the MAD of the test set from 0.192 to 0.097 eV (R2 = 0.833).
中文翻译:

精确的电离势量子化学计算:通过实验和基准量子化学计算获得的大量数据验证DFT-LOC方法的有效性
已知密度函数理论(DFT)在计算热力学值(例如电离势(IPs))时通常会由于非动力学误差(即自相互作用项)而失败。通过为原子轨道,键以及成对和不成对的电子分配相应的校正而得出的局部轨道校正(LOC)用于校正根据DFT计算的IP。一些分配的参数,实际上是由于键周围环境的收缩和变化而导致的,取决于使用中性和氧化性物质之间电荷密度的差异来确定分子中要除去电子的位置。在我们的训练集中,使用NIST数据库收集了文献中报告的实验IP的各种有机和无机小分子。对于不确定或没有实验测量值的某些分子,我们使用耦合簇理论和辅助场量子蒙特卡洛方法获得IP。应用最小二乘回归生成的这些校正后,LOC将训练集的平均绝对偏差(MAD)从0.143降低至0.046 eV(R 2 = 0.895),并且LOC将测试集的MAD从0.192降至0.097 eV(R 2 = 0.833)。
更新日期:2020-04-24
中文翻译:
精确的电离势量子化学计算:通过实验和基准量子化学计算获得的大量数据验证DFT-LOC方法的有效性
已知密度函数理论(DFT)在计算热力学值(例如电离势(IPs))时通常会由于非动力学误差(即自相互作用项)而失败。通过为原子轨道,键以及成对和不成对的电子分配相应的校正而得出的局部轨道校正(LOC)用于校正根据DFT计算的IP。一些分配的参数,实际上是由于键周围环境的收缩和变化而导致的,取决于使用中性和氧化性物质之间电荷密度的差异来确定分子中要除去电子的位置。在我们的训练集中,使用NIST数据库收集了文献中报告的实验IP的各种有机和无机小分子。对于不确定或没有实验测量值的某些分子,我们使用耦合簇理论和辅助场量子蒙特卡洛方法获得IP。应用最小二乘回归生成的这些校正后,LOC将训练集的平均绝对偏差(MAD)从0.143降低至0.046 eV(R 2 = 0.895),并且LOC将测试集的MAD从0.192降至0.097 eV(R 2 = 0.833)。










































 京公网安备 11010802027423号
京公网安备 11010802027423号