当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Prediction of protein–protein complexes using replica exchange with repulsive scaling
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-03-09 , DOI: 10.1002/jcc.26187 Till Siebenmorgen 1 , Michael Engelhard 1 , Martin Zacharias 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-03-09 , DOI: 10.1002/jcc.26187 Till Siebenmorgen 1 , Michael Engelhard 1 , Martin Zacharias 1
Affiliation
|
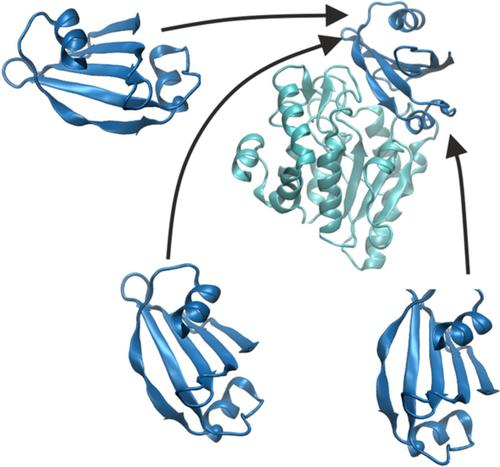
The realistic prediction of protein–protein complex structures is import to ultimately model the interaction of all proteins in a cell and for the design of new protein–protein interactions. In principle, molecular dynamics (MD) simulations allow one to follow the association process under realistic conditions including full partner flexibility and surrounding solvent. However, due to the many local binding energy minima at the surface of protein partners, MD simulations are frequently trapped for long times in transient association states. We have designed a replica‐exchange based scheme employing different levels of a repulsive biasing between partners in each replica simulation. The bias acts only on intermolecular interactions based on an increase in effective pairwise van der Waals radii (repulsive scaling (RS)‐REMD) without affecting interactions within each protein or with the solvent. For a set of five protein test cases (out of six) the RS‐REMD technique allowed the sampling of near‐native complex structures even when starting from the opposide site with respect to the native binding site for one partner. Using the same start structures and same computational demand regular MD simulations sampled near native complex structures only for one case. The method showed also improved results for the refinement of docked structures in the vicinity of the native binding geometry compared to regular MD refinement.
中文翻译:

使用具有排斥缩放的复制品交换预测蛋白质-蛋白质复合物
蛋白质-蛋白质复合物结构的现实预测对于最终模拟细胞中所有蛋白质的相互作用以及设计新的蛋白质-蛋白质相互作用非常重要。原则上,分子动力学 (MD) 模拟允许人们在现实条件下跟踪关联过程,包括完全合作伙伴的灵活性和周围的溶剂。然而,由于蛋白质伴侣表面的许多局部结合能最小值,MD 模拟经常被困在瞬态关联状态中很长时间。我们设计了一个基于副本交换的方案,在每个副本模拟中采用不同级别的合作伙伴之间的排斥偏差。偏差仅作用于基于有效成对范德华半径(排斥缩放(RS)-REMD)增加的分子间相互作用,而不影响每个蛋白质内或与溶剂的相互作用。对于一组五个蛋白质测试案例(六个中),RS-REMD 技术允许对接近原生的复杂结构进行采样,即使是从相对于一个伴侣的天然结合位点的对侧位点开始也是如此。使用相同的起始结构和相同的计算需求,常规 MD 模拟仅针对一种情况在本地复杂结构附近采样。与常规 MD 细化相比,该方法还显示了改进天然结合几何附近对接结构的改进结果。对于一组五个蛋白质测试案例(六个中),RS-REMD 技术允许对接近原生的复杂结构进行采样,即使是从相对于一个伴侣的天然结合位点的对侧位点开始也是如此。使用相同的起始结构和相同的计算需求,常规 MD 模拟仅针对一种情况在本地复杂结构附近采样。与常规 MD 细化相比,该方法还显示了改进天然结合几何附近对接结构的改进结果。对于一组五个蛋白质测试案例(六个中),RS-REMD 技术允许对接近原生的复杂结构进行采样,即使是从相对于一个伴侣的天然结合位点的对侧位点开始也是如此。使用相同的起始结构和相同的计算需求,常规 MD 模拟仅针对一种情况在本地复杂结构附近采样。与常规 MD 细化相比,该方法还显示了改进天然结合几何附近对接结构的改进结果。
更新日期:2020-03-09
中文翻译:
使用具有排斥缩放的复制品交换预测蛋白质-蛋白质复合物
蛋白质-蛋白质复合物结构的现实预测对于最终模拟细胞中所有蛋白质的相互作用以及设计新的蛋白质-蛋白质相互作用非常重要。原则上,分子动力学 (MD) 模拟允许人们在现实条件下跟踪关联过程,包括完全合作伙伴的灵活性和周围的溶剂。然而,由于蛋白质伴侣表面的许多局部结合能最小值,MD 模拟经常被困在瞬态关联状态中很长时间。我们设计了一个基于副本交换的方案,在每个副本模拟中采用不同级别的合作伙伴之间的排斥偏差。偏差仅作用于基于有效成对范德华半径(排斥缩放(RS)-REMD)增加的分子间相互作用,而不影响每个蛋白质内或与溶剂的相互作用。对于一组五个蛋白质测试案例(六个中),RS-REMD 技术允许对接近原生的复杂结构进行采样,即使是从相对于一个伴侣的天然结合位点的对侧位点开始也是如此。使用相同的起始结构和相同的计算需求,常规 MD 模拟仅针对一种情况在本地复杂结构附近采样。与常规 MD 细化相比,该方法还显示了改进天然结合几何附近对接结构的改进结果。对于一组五个蛋白质测试案例(六个中),RS-REMD 技术允许对接近原生的复杂结构进行采样,即使是从相对于一个伴侣的天然结合位点的对侧位点开始也是如此。使用相同的起始结构和相同的计算需求,常规 MD 模拟仅针对一种情况在本地复杂结构附近采样。与常规 MD 细化相比,该方法还显示了改进天然结合几何附近对接结构的改进结果。对于一组五个蛋白质测试案例(六个中),RS-REMD 技术允许对接近原生的复杂结构进行采样,即使是从相对于一个伴侣的天然结合位点的对侧位点开始也是如此。使用相同的起始结构和相同的计算需求,常规 MD 模拟仅针对一种情况在本地复杂结构附近采样。与常规 MD 细化相比,该方法还显示了改进天然结合几何附近对接结构的改进结果。










































 京公网安备 11010802027423号
京公网安备 11010802027423号