当前位置:
X-MOL 学术
›
Nat. Microbiol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Spatially distinct physiology of Bacteroides fragilis within the proximal colon of gnotobiotic mice.
Nature Microbiology ( IF 20.5 ) Pub Date : 2020-03-09 , DOI: 10.1038/s41564-020-0683-3 Gregory P Donaldson 1 , Wen-Chi Chou 2 , Abigail L Manson 2 , Peter Rogov 2 , Thomas Abeel 2, 3 , James Bochicchio 2 , Dawn Ciulla 2 , Alexandre Melnikov 2 , Peter B Ernst 4 , Hiutung Chu 4 , Georgia Giannoukos 2 , Ashlee M Earl 2 , Sarkis K Mazmanian 1
Nature Microbiology ( IF 20.5 ) Pub Date : 2020-03-09 , DOI: 10.1038/s41564-020-0683-3 Gregory P Donaldson 1 , Wen-Chi Chou 2 , Abigail L Manson 2 , Peter Rogov 2 , Thomas Abeel 2, 3 , James Bochicchio 2 , Dawn Ciulla 2 , Alexandre Melnikov 2 , Peter B Ernst 4 , Hiutung Chu 4 , Georgia Giannoukos 2 , Ashlee M Earl 2 , Sarkis K Mazmanian 1
Affiliation
|
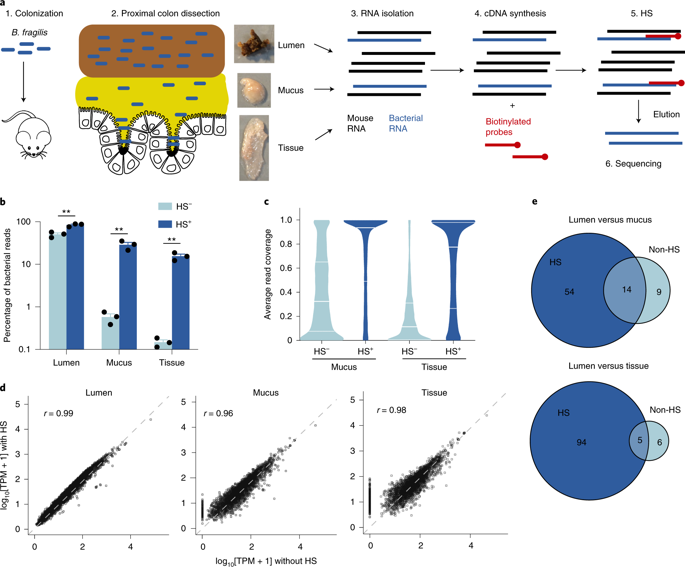
A complex microbiota inhabits various microenvironments of the gut, with some symbiotic bacteria having evolved traits to invade the epithelial mucus layer and reside deep within the intestinal tissue of animals. Whether these distinct bacterial communities across gut biogeographies exhibit divergent behaviours is largely unknown. Global transcriptomic analysis to investigate microbial physiology in specific mucosal niches has been hampered technically by an overabundance of host RNA. Here, we employed hybrid selection RNA sequencing (hsRNA-Seq) to enable detailed spatial transcriptomic profiling of a prominent human commensal as it colonizes the colonic lumen, mucus or epithelial tissue of mice. Compared to conventional RNA-Seq, hsRNA-Seq increased reads mapping to the Bacteroides fragilis genome by 48- and 154-fold in mucus and tissue, respectively, allowing for high-fidelity comparisons across biogeographic sites. Near the epithelium, B. fragilis upregulated numerous genes involved in protein synthesis, indicating that bacteria inhabiting the mucosal niche are metabolically active. Further, a specific sulfatase (BF3086) and glycosyl hydrolase (BF3134) were highly induced in mucus and tissue compared to bacteria in the lumen. In-frame deletion of these genes impaired in vitro growth on mucus as a carbon source, as well as mucosal colonization of mice. Mutants in either B. fragilis gene displayed a fitness defect in competing for colonization against bacterial challenge, revealing the importance of site-specific gene expression for robust host-microbial symbiosis. As a versatile tool, hsRNA-Seq can be deployed to explore the in vivo spatial physiology of numerous bacterial pathogens or commensals.
中文翻译:

知生小鼠近端结肠内脆弱拟杆菌的空间不同生理学。
复杂的微生物群栖息在肠道的各种微环境中,一些共生细菌进化出侵入上皮粘液层并深入动物肠道组织的特征。肠道生物地理学中这些不同的细菌群落是否表现出不同的行为在很大程度上是未知的。用于研究特定粘膜生态位中微生物生理学的全球转录组分析在技术上受到宿主 RNA 过多的阻碍。在这里,我们采用杂交选择 RNA 测序 (hsRNA-Seq) 来对显着的人类共生体进行详细的空间转录组分析,因为它定植于小鼠的结肠腔、粘液或上皮组织。与传统的 RNA-Seq 相比,hsRNA-Seq 在粘液和组织中将比对到脆弱拟杆菌基因组的读数增加了 48 倍和 154 倍,分别允许跨生物地理站点进行高保真比较。在上皮附近,脆弱拟杆菌上调了许多参与蛋白质合成的基因,这表明居住在粘膜生态位的细菌代谢活跃。此外,与管腔中的细菌相比,粘液和组织中的特异性硫酸酯酶 (BF3086) 和糖基水解酶 (BF3134) 被高度诱导。这些基因的框内缺失损害了作为碳源的粘液的体外生长,以及小鼠的粘膜定植。B. fragilis 基因中的突变体在与细菌挑战竞争定植时表现出适应性缺陷,揭示了位点特异性基因表达对于强大的宿主-微生物共生的重要性。作为一种多功能工具,
更新日期:2020-03-09
中文翻译:
知生小鼠近端结肠内脆弱拟杆菌的空间不同生理学。
复杂的微生物群栖息在肠道的各种微环境中,一些共生细菌进化出侵入上皮粘液层并深入动物肠道组织的特征。肠道生物地理学中这些不同的细菌群落是否表现出不同的行为在很大程度上是未知的。用于研究特定粘膜生态位中微生物生理学的全球转录组分析在技术上受到宿主 RNA 过多的阻碍。在这里,我们采用杂交选择 RNA 测序 (hsRNA-Seq) 来对显着的人类共生体进行详细的空间转录组分析,因为它定植于小鼠的结肠腔、粘液或上皮组织。与传统的 RNA-Seq 相比,hsRNA-Seq 在粘液和组织中将比对到脆弱拟杆菌基因组的读数增加了 48 倍和 154 倍,分别允许跨生物地理站点进行高保真比较。在上皮附近,脆弱拟杆菌上调了许多参与蛋白质合成的基因,这表明居住在粘膜生态位的细菌代谢活跃。此外,与管腔中的细菌相比,粘液和组织中的特异性硫酸酯酶 (BF3086) 和糖基水解酶 (BF3134) 被高度诱导。这些基因的框内缺失损害了作为碳源的粘液的体外生长,以及小鼠的粘膜定植。B. fragilis 基因中的突变体在与细菌挑战竞争定植时表现出适应性缺陷,揭示了位点特异性基因表达对于强大的宿主-微生物共生的重要性。作为一种多功能工具,










































 京公网安备 11010802027423号
京公网安备 11010802027423号