Biochimica et Biophysica Acta (BBA) - Biomembranes ( IF 2.8 ) Pub Date : 2020-03-07 , DOI: 10.1016/j.bbamem.2020.183265 Bhagyashree D Rao 1 , Parijat Sarkar 2 , Amitabha Chattopadhyay 3
|
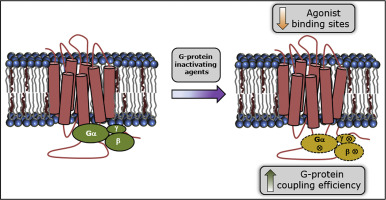
G protein-coupled receptors (GPCRs) constitute the largest superfamily of membrane proteins in higher eukaryotes, and facilitate information transfer from the extracellular environment to the cellular interior upon activation by ligands. Their role in diverse signaling processes makes them an attractive choice as drug targets. GPCRs are coupled to heterotrimeric G-proteins which represent an important interface through which signal transduction occurs across the plasma membrane upon activation by ligands. To obtain further insight into the molecular details of interaction of G-proteins with GPCRs, in this work, we explored the selectivity of binding of specific agonists and antagonists to the serotonin1A receptor under conditions of progressive G-protein inactivation. The serotonin1A receptor is an important neurotransmitter receptor belonging to the GPCR family and is a popular drug target. By use of a number of agents to inactivate G-proteins, we show here that the serotonin1A receptor displays differential discrimination between agonist and antagonist binding. Our results show a reduction in binding sites of the receptor upon treatment with G-protein inactivating agents. In addition, G-protein coupling efficiency was enhanced when G-proteins were inactivated using urea and alkaline pH. We envision that our results could be useful in achieving multiple signaling states of the receptor by fine tuning the conditions of G-protein inactivation and in structural biology of GPCRs bound to specific ligands.
中文翻译:
通过G蛋白偶联与Serotonin1A受体结合的激动剂和拮抗剂的选择性。
G蛋白偶联受体(GPCR)构成了高等真核生物中最大的膜蛋白超家族,并在配体激活后促进信息从细胞外环境转移到细胞内部。它们在多种信号传导过程中的作用使其成为药物靶标的诱人选择。GPCR与异三聚体G蛋白偶联,异三聚体G蛋白代表重要的界面,通过配体激活,信号通过该界面跨质膜发生。为了进一步了解G蛋白与GPCR相互作用的分子细节,在这项工作中,我们探索了在逐步G蛋白失活条件下特异性激动剂和拮抗剂与血清素1A受体结合的选择性。血清素1A受体是属于GPCR家族的重要神经递质受体,并且是受欢迎的药物靶标。通过使用多种试剂来失活G蛋白,我们在这里表明,血清素1A受体在激动剂和拮抗剂结合之间表现出不同的区别。我们的结果表明,用G蛋白灭活剂处理后,受体结合位点减少。此外,当使用尿素和碱性pH灭活G蛋白时,G蛋白偶联效率会提高。我们设想,我们的结果可通过微调G蛋白失活的条件以及结合特定配体的GPCR的结构生物学来实现受体的多种信号状态。










































 京公网安备 11010802027423号
京公网安备 11010802027423号