当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
STACKED - Solvation Theory of Aromatic Complexes as Key for Estimating Drug Binding.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-06 , DOI: 10.1021/acs.jcim.9b01165 Johannes R Loeffler 1 , Monica L Fernández-Quintero 1 , Michael Schauperl 1 , Klaus R Liedl 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-06 , DOI: 10.1021/acs.jcim.9b01165 Johannes R Loeffler 1 , Monica L Fernández-Quintero 1 , Michael Schauperl 1 , Klaus R Liedl 1
Affiliation
|
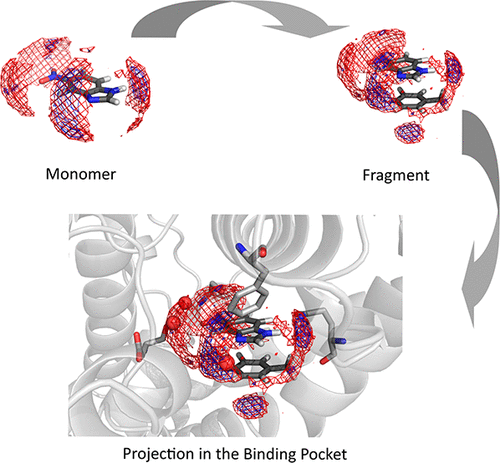
The use of fragments to biophysically characterize a protein binding pocket and determine the strengths of certain interactions is a computationally and experimentally commonly applied approach. Almost all drug like molecules contain at least one aromatic moiety forming stacking interactions in the binding pocket. In computational drug design, the strength of stacking and the resulting optimization of the aromatic core or moiety is usually calculated using high level quantum mechanical approaches. However, as these calculations are performed in a vacuum, solvation properties are neglected. We close this gap by using Grid Inhomogeneous Solvation Theory (GIST) to describe the properties of individual heteroaromatics and complexes and thereby estimate the desolvation penalty. In our study, we investigated the solvation free energies of heteroaromatics frequently occurring in drug design projects in complex with truncated side chains of phenylalanine, tyrosine, and tryptophan. Furthermore, we investigated the properties of drug-fragments crystallized in a fragment-based lead optimization approach investigating PDE-10-A. We do not only find good correlation for the estimated desolvation penalty and the experimental binding free energy, but our calculations also allow us to predict prominent interaction sites. We highlight the importance of including the desolvation penalty of the respective heteroaromatics in stacked complexes to explain the gain or loss in affinity of potential lead compounds.
中文翻译:

堆叠-芳香族化合物的溶剂化理论是估计药物结合力的关键。
使用片段进行生物物理学上的表征蛋白结合袋并确定某些相互作用的强度是计算和实验上普遍采用的方法。几乎所有药物样分子都包含至少一个在结合袋中形成堆积相互作用的芳族部分。在药物的计算设计中,通常使用高级量子力学方法来计算堆积强度以及所得到的芳族核或芳基部分的优化。但是,由于这些计算是在真空中进行的,因此溶剂化性能被忽略。我们通过使用网格非均匀溶剂化理论(GIST)来描述单个杂芳族化合物和配合物的性质,从而估计去溶剂化损失,从而弥合了这一差距。在我们的研究中 我们研究了在药物设计项目中经常发生的杂芳族化合物的溶剂化自由能,其中苯丙氨酸,酪氨酸和色氨酸的侧链被截断。此外,我们研究了在研究PDE-10-A的基于片段的铅优化方法中结晶的药物片段的性质。我们不仅发现估计的去溶剂化罚分与实验的结合自由能之间具有良好的相关性,而且我们的计算还使我们能够预测突出的相互作用位点。我们强调了在堆叠复合物中包括各个杂芳族化合物的去溶剂化罚金的重要性,以解释潜在铅化合物亲和力的获得或损失。我们研究了在研究PDE-10-A的基于片段的铅优化方法中结晶的药物片段的性质。我们不仅发现了估计的去溶剂化罚分与实验性结合自由能之间具有良好的相关性,而且我们的计算还使我们能够预测出显着的相互作用位点。我们强调了在堆叠复合物中包括各个杂芳族化合物的去溶剂化罚金的重要性,以解释潜在铅化合物亲和力的获得或损失。我们研究了在研究PDE-10-A的基于片段的铅优化方法中结晶的药物片段的性质。我们不仅发现了估计的去溶剂化罚分与实验性结合自由能之间具有良好的相关性,而且我们的计算还使我们能够预测出显着的相互作用位点。我们强调了在堆叠复合物中包括各个杂芳族化合物的去溶剂化罚金的重要性,以解释潜在铅化合物亲和力的获得或损失。
更新日期:2020-03-06
中文翻译:
堆叠-芳香族化合物的溶剂化理论是估计药物结合力的关键。
使用片段进行生物物理学上的表征蛋白结合袋并确定某些相互作用的强度是计算和实验上普遍采用的方法。几乎所有药物样分子都包含至少一个在结合袋中形成堆积相互作用的芳族部分。在药物的计算设计中,通常使用高级量子力学方法来计算堆积强度以及所得到的芳族核或芳基部分的优化。但是,由于这些计算是在真空中进行的,因此溶剂化性能被忽略。我们通过使用网格非均匀溶剂化理论(GIST)来描述单个杂芳族化合物和配合物的性质,从而估计去溶剂化损失,从而弥合了这一差距。在我们的研究中 我们研究了在药物设计项目中经常发生的杂芳族化合物的溶剂化自由能,其中苯丙氨酸,酪氨酸和色氨酸的侧链被截断。此外,我们研究了在研究PDE-10-A的基于片段的铅优化方法中结晶的药物片段的性质。我们不仅发现估计的去溶剂化罚分与实验的结合自由能之间具有良好的相关性,而且我们的计算还使我们能够预测突出的相互作用位点。我们强调了在堆叠复合物中包括各个杂芳族化合物的去溶剂化罚金的重要性,以解释潜在铅化合物亲和力的获得或损失。我们研究了在研究PDE-10-A的基于片段的铅优化方法中结晶的药物片段的性质。我们不仅发现了估计的去溶剂化罚分与实验性结合自由能之间具有良好的相关性,而且我们的计算还使我们能够预测出显着的相互作用位点。我们强调了在堆叠复合物中包括各个杂芳族化合物的去溶剂化罚金的重要性,以解释潜在铅化合物亲和力的获得或损失。我们研究了在研究PDE-10-A的基于片段的铅优化方法中结晶的药物片段的性质。我们不仅发现了估计的去溶剂化罚分与实验性结合自由能之间具有良好的相关性,而且我们的计算还使我们能够预测出显着的相互作用位点。我们强调了在堆叠复合物中包括各个杂芳族化合物的去溶剂化罚金的重要性,以解释潜在铅化合物亲和力的获得或损失。










































 京公网安备 11010802027423号
京公网安备 11010802027423号