当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A Grid Map Based Approach to Identify Nonobvious Ligand Design Opportunities in 3D Protein Structure Ensembles.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-05 , DOI: 10.1021/acs.jcim.0c00051 Philipp S Schmalhorst 1 , Andreas Bergner 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-05 , DOI: 10.1021/acs.jcim.0c00051 Philipp S Schmalhorst 1 , Andreas Bergner 1
Affiliation
|
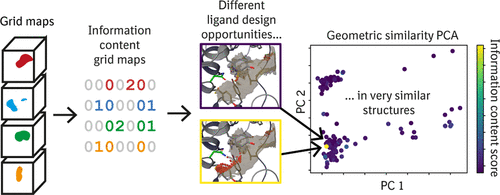
Three-dimensional protein structures are a key requisite for structure-based drug discovery. For many highly relevant targets, medicinal chemists are confronted with large numbers of target structures in their apo-forms or in complex with a wealth of different ligands. To exploit the full potential of such structure ensembles, in terms of aggregated knowledge that informs design, it is desirable to extract a manageable number of structures that provide a maximum of ligand design opportunities. Most commonly used structure comparison methods are largely based on atom positions and geometry-based metrics; medicinal chemists, however, seek ligand design opportunities and are interested in methods that allow such information to be distilled from structural data and guide them in an intuitive way. Here we present an approach for identifying nonobvious ligand design opportunities in protein conformation ensembles based on the information content in grid maps that represent, for example, binding hotspots. We use four different examples to show how this method can provide information orthogonal to established coordinate-based similarity methods. Furthermore, we demonstrate that ligand design opportunities can change substantially with very small structural variations. We expect that this approach will advance the identification of ligand design opportunities hidden in large collections of protein-ligand complex data that would otherwise have been missed.
中文翻译:

一种基于网格图的方法来识别3D蛋白质结构集合体中的非明显配体设计机会。
三维蛋白质结构是基于结构的药物发现的关键条件。对于许多高度相关的靶标,药用化学家们面临着许多其脱辅基形式或与大量不同配体复合的靶标结构。为了充分利用这种结构集合体的全部潜力,就有助于设计的汇总知识而言,希望提取出可管理数量的结构,从而提供最大的配体设计机会。最常用的结构比较方法主要基于原子位置和基于几何的度量。然而,药用化学家寻求配体设计的机会,并且对允许从结构数据中提取此类信息并以直观方式进行指导的方法感兴趣。在这里,我们提出了一种方法,用于根据代表例如结合热点的网格图中的信息内容,确定蛋白质构象集合中非显而易见的配体设计机会。我们使用四个不同的示例来说明此方法如何提供与已建立的基于坐标的相似性方法正交的信息。此外,我们证明配体的设计机会可以通过非常小的结构变化而显着改变。我们期望这种方法将促进鉴定隐藏在大量蛋白质-配体复杂数据中的配体设计机会,而这些数据否则会被遗漏。我们使用四个不同的示例来说明此方法如何提供与已建立的基于坐标的相似性方法正交的信息。此外,我们证明配体的设计机会可以通过非常小的结构变化而显着改变。我们期望这种方法将促进鉴定隐藏在大量蛋白质-配体复杂数据中的配体设计机会,而这些数据否则会被遗漏。我们使用四个不同的示例来说明此方法如何提供与已建立的基于坐标的相似性方法正交的信息。此外,我们证明配体的设计机会可以通过非常小的结构变化而显着改变。我们期望这种方法将促进鉴定隐藏在大量蛋白质-配体复杂数据中的配体设计机会,而这些数据否则会被遗漏。
更新日期:2020-03-05
中文翻译:
一种基于网格图的方法来识别3D蛋白质结构集合体中的非明显配体设计机会。
三维蛋白质结构是基于结构的药物发现的关键条件。对于许多高度相关的靶标,药用化学家们面临着许多其脱辅基形式或与大量不同配体复合的靶标结构。为了充分利用这种结构集合体的全部潜力,就有助于设计的汇总知识而言,希望提取出可管理数量的结构,从而提供最大的配体设计机会。最常用的结构比较方法主要基于原子位置和基于几何的度量。然而,药用化学家寻求配体设计的机会,并且对允许从结构数据中提取此类信息并以直观方式进行指导的方法感兴趣。在这里,我们提出了一种方法,用于根据代表例如结合热点的网格图中的信息内容,确定蛋白质构象集合中非显而易见的配体设计机会。我们使用四个不同的示例来说明此方法如何提供与已建立的基于坐标的相似性方法正交的信息。此外,我们证明配体的设计机会可以通过非常小的结构变化而显着改变。我们期望这种方法将促进鉴定隐藏在大量蛋白质-配体复杂数据中的配体设计机会,而这些数据否则会被遗漏。我们使用四个不同的示例来说明此方法如何提供与已建立的基于坐标的相似性方法正交的信息。此外,我们证明配体的设计机会可以通过非常小的结构变化而显着改变。我们期望这种方法将促进鉴定隐藏在大量蛋白质-配体复杂数据中的配体设计机会,而这些数据否则会被遗漏。我们使用四个不同的示例来说明此方法如何提供与已建立的基于坐标的相似性方法正交的信息。此外,我们证明配体的设计机会可以通过非常小的结构变化而显着改变。我们期望这种方法将促进鉴定隐藏在大量蛋白质-配体复杂数据中的配体设计机会,而这些数据否则会被遗漏。


























 京公网安备 11010802027423号
京公网安备 11010802027423号