当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Density Functional Computations of Vibrational Circular Dichroism Spectra beyond the Born–Oppenheimer Approximation
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-05 , DOI: 10.1021/acs.jctc.0c00081 Josef Tomeček 1 , Petr Bouř 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-05 , DOI: 10.1021/acs.jctc.0c00081 Josef Tomeček 1 , Petr Bouř 2
Affiliation
|
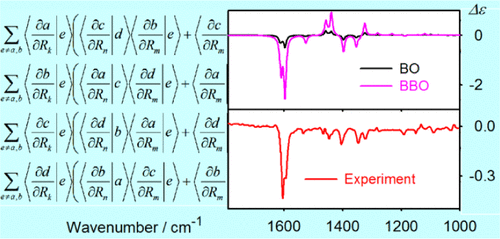
Transition-metal complexes provide rich features in vibrational circular dichroism (VCD) spectra, including significant intensity enhancements, and become thus useful in structural and functional studies of molecules. Quite often, however, the vibrational spectral bands are mixed with the electronic ones, and interpretation of such experiments is difficult. In the present study, we elaborate on the theory needed to calculate the VCD intensities beyond the Born–Oppenheimer (BO) approximation. Within a perturbation approach, the coupling between the electronic and vibrational states is estimated using the harmonic approximation and simplified wave functions obtainable from common density functional theory (DFT) computations. Explicit expressions, including Slater determinants and derivatives of molecular orbitals, are given. On a model diamine complex, the implementation is tested and factors affecting spectral intensities and frequencies are investigated. For two larger molecules, the results are in a qualitative agreement with previous experimental data. Typically, the electronic–vibrational interaction Hamiltonian coupling elements are rather small (∼0 to 10 cm–1), which provides negligible contributions to vibrational frequencies and absorption intensities. However, significant changes in VCD spectra are induced due to the large transition magnetic dipole moment associated with the d–d metal transitions. The possibility to model the spectra beyond the BO limit opens the way to further applications of chiral spectroscopy and transition-metal complexes.
中文翻译:

超越Born–Oppenheimer逼近的振动圆二向色光谱的密度泛函计算
过渡金属配合物在振动圆二色性(VCD)光谱中提供了丰富的功能,包括明显的强度增强,因此可用于分子的结构和功能研究。然而,振动光谱带经常与电子频带混合,并且难以解释这种实验。在本研究中,我们将详细阐述计算超出Born–Oppenheimer(BO)近似值的VCD强度所需的理论。在微扰方法中,电子和振动状态之间的耦合是使用谐波近似和简化波函数来估计的,而波函数可以从通用密度泛函理论(DFT)计算中获得。给出了明确的表达式,包括Slater行列式和分子轨道的导数。在模型二胺络合物上,测试了其实现方式,并研究了影响光谱强度和频率的因素。对于两个较大的分子,结果与先前的实验数据在质量上一致。通常,电子振动相互作用的哈密顿耦合元素很小(〜0至10 cm–1),它对振动频率和吸收强度的贡献可忽略不计。但是,由于与d–d金属跃迁相关的大跃迁磁偶极矩,导致VCD光谱发生了显着变化。对超出BO限制的光谱进行建模的可能性为手性光谱学和过渡金属配合物的进一步应用开辟了道路。
更新日期:2020-04-24
中文翻译:
超越Born–Oppenheimer逼近的振动圆二向色光谱的密度泛函计算
过渡金属配合物在振动圆二色性(VCD)光谱中提供了丰富的功能,包括明显的强度增强,因此可用于分子的结构和功能研究。然而,振动光谱带经常与电子频带混合,并且难以解释这种实验。在本研究中,我们将详细阐述计算超出Born–Oppenheimer(BO)近似值的VCD强度所需的理论。在微扰方法中,电子和振动状态之间的耦合是使用谐波近似和简化波函数来估计的,而波函数可以从通用密度泛函理论(DFT)计算中获得。给出了明确的表达式,包括Slater行列式和分子轨道的导数。在模型二胺络合物上,测试了其实现方式,并研究了影响光谱强度和频率的因素。对于两个较大的分子,结果与先前的实验数据在质量上一致。通常,电子振动相互作用的哈密顿耦合元素很小(〜0至10 cm–1),它对振动频率和吸收强度的贡献可忽略不计。但是,由于与d–d金属跃迁相关的大跃迁磁偶极矩,导致VCD光谱发生了显着变化。对超出BO限制的光谱进行建模的可能性为手性光谱学和过渡金属配合物的进一步应用开辟了道路。










































 京公网安备 11010802027423号
京公网安备 11010802027423号