当前位置:
X-MOL 学术
›
Clin. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Report of the first patient with a homozygous OTUD7A variant responsible for epileptic encephalopathy and related proteasome dysfunction.
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-02-11 , DOI: 10.1111/cge.13709 Philippine Garret 1, 2 , Frédéric Ebstein 3 , Geoffroy Delplancq 1, 4 , Blandine Dozieres-Puyravel 5 , Aïcha Boughalem 2 , Stéphane Auvin 5, 6 , Yannis Duffourd 1, 4 , Sandro Klafack 3 , Barbara A Zieba 3 , Sana Mahmoudi 7 , Karun K Singh 8 , Laurence Duplomb 1, 4 , Christel Thauvin-Robinet 1, 4, 9 , Jean-Marc Costa 2 , Elke Krüger 3 , Detlef Trost 2 , Alain Verloes 6, 10 , Laurence Faivre 1, 11 , Antonio Vitobello 1, 4
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-02-11 , DOI: 10.1111/cge.13709 Philippine Garret 1, 2 , Frédéric Ebstein 3 , Geoffroy Delplancq 1, 4 , Blandine Dozieres-Puyravel 5 , Aïcha Boughalem 2 , Stéphane Auvin 5, 6 , Yannis Duffourd 1, 4 , Sandro Klafack 3 , Barbara A Zieba 3 , Sana Mahmoudi 7 , Karun K Singh 8 , Laurence Duplomb 1, 4 , Christel Thauvin-Robinet 1, 4, 9 , Jean-Marc Costa 2 , Elke Krüger 3 , Detlef Trost 2 , Alain Verloes 6, 10 , Laurence Faivre 1, 11 , Antonio Vitobello 1, 4
Affiliation
|
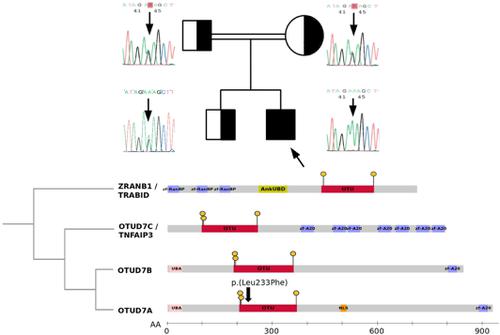
Heterozygous microdeletions of chromosome 15q13.3 (MIM: 612001) show incomplete penetrance and are associated with a highly variable phenotype that may include intellectual disability, epilepsy, facial dysmorphism and digit anomalies. Rare patients carrying homozygous deletions show more severe phenotypes including epileptic encephalopathy, hypotonia and poor growth. For years, CHRNA7 (MIM: 118511), was considered the candidate gene that could account for this syndrome. However, recent studies in mouse models have shown that OTUD7A/CEZANNE2 (MIM: 612024), which encodes for an ovarian tumor (OTU) deubiquitinase, should be considered the critical gene responsible for brain dysfunction. In this study, a patient presenting with severe global developmental delay, language impairment and epileptic encephalopathy was referred to our genetics center. Trio exome sequencing (tES) analysis identified a homozygous OTUD7A missense variant (NM_130901.2:c.697C>T), predicted to alter an ultraconserved amino acid, p.(Leu233Phe), lying within the OTU catalytic domain. Its subsequent segregation analysis revealed that the parents, presenting with learning disability, and brother were heterozygous carriers. Biochemical assays demonstrated that proteasome complex formation and function were significantly reduced in patient-derived fibroblasts and in OTUD7A knockout HAP1 cell line. We provide evidence that biallelic pathogenic OTUD7A variation is linked to early-onset epileptic encephalopathy and proteasome dysfunction.
中文翻译:

第一位患有纯合性OTUD7A变异体的患者的报告,该变异体引起癫痫性脑病和相关的蛋白酶体功能障碍。
染色体15q13.3(MIM:612001)的杂合微缺失显示出不完全的外显率,并与高度可变的表型有关,该表型可能包括智力残疾,癫痫,面部畸形和手指异常。罕见的携带纯合子缺失的患者表现出更严重的表型,包括癫痫性脑病,肌张力低下和生长不良。多年来,CHRNA7(MIM:118511)被视为可能导致该综合征的候选基因。但是,最近在小鼠模型中的研究表明,编码卵巢肿瘤(OTU)去泛素酶的OTUD7A / CEZANNE2(MIM:612024)应该被认为是导致脑功能异常的关键基因。在这项研究中,出现严重的整体发育迟缓,语言障碍和癫痫性脑病的患者被转介到我们的遗传学中心。三重基因组外显子测序(tES)分析确定了纯合的OTUD7A错义变体(NM_130901.2:c.697C> T),预计会改变OTU催化域内的超保守氨基酸p。(Leu233Phe)。其随后的隔离分析显示,存在学习障碍的父母和兄弟是杂合子携带者。生化分析表明,在患者来源的成纤维细胞和OTUD7A敲除的HAP1细胞系中,蛋白酶体复合物的形成和功能显着降低。我们提供证据,双等位基因致病性OTUD7A变异与早期发作的癫痫性脑病和蛋白酶体功能障碍有关。位于OTU催化域内。其随后的隔离分析显示,存在学习障碍的父母和兄弟是杂合子携带者。生化分析表明,在患者来源的成纤维细胞和OTUD7A敲除的HAP1细胞系中,蛋白酶体复合物的形成和功能显着降低。我们提供证据,双等位基因致病性OTUD7A变异与早期发作的癫痫性脑病和蛋白酶体功能障碍有关。位于OTU催化域内。其随后的隔离分析显示,存在学习障碍的父母和兄弟是杂合子携带者。生化分析表明,在患者来源的成纤维细胞和OTUD7A敲除的HAP1细胞系中,蛋白酶体复合物的形成和功能显着降低。我们提供证据,双等位基因致病性OTUD7A变异与早发性癫痫性脑病和蛋白酶体功能障碍有关。
更新日期:2020-03-26
中文翻译:
第一位患有纯合性OTUD7A变异体的患者的报告,该变异体引起癫痫性脑病和相关的蛋白酶体功能障碍。
染色体15q13.3(MIM:612001)的杂合微缺失显示出不完全的外显率,并与高度可变的表型有关,该表型可能包括智力残疾,癫痫,面部畸形和手指异常。罕见的携带纯合子缺失的患者表现出更严重的表型,包括癫痫性脑病,肌张力低下和生长不良。多年来,CHRNA7(MIM:118511)被视为可能导致该综合征的候选基因。但是,最近在小鼠模型中的研究表明,编码卵巢肿瘤(OTU)去泛素酶的OTUD7A / CEZANNE2(MIM:612024)应该被认为是导致脑功能异常的关键基因。在这项研究中,出现严重的整体发育迟缓,语言障碍和癫痫性脑病的患者被转介到我们的遗传学中心。三重基因组外显子测序(tES)分析确定了纯合的OTUD7A错义变体(NM_130901.2:c.697C> T),预计会改变OTU催化域内的超保守氨基酸p。(Leu233Phe)。其随后的隔离分析显示,存在学习障碍的父母和兄弟是杂合子携带者。生化分析表明,在患者来源的成纤维细胞和OTUD7A敲除的HAP1细胞系中,蛋白酶体复合物的形成和功能显着降低。我们提供证据,双等位基因致病性OTUD7A变异与早期发作的癫痫性脑病和蛋白酶体功能障碍有关。位于OTU催化域内。其随后的隔离分析显示,存在学习障碍的父母和兄弟是杂合子携带者。生化分析表明,在患者来源的成纤维细胞和OTUD7A敲除的HAP1细胞系中,蛋白酶体复合物的形成和功能显着降低。我们提供证据,双等位基因致病性OTUD7A变异与早期发作的癫痫性脑病和蛋白酶体功能障碍有关。位于OTU催化域内。其随后的隔离分析显示,存在学习障碍的父母和兄弟是杂合子携带者。生化分析表明,在患者来源的成纤维细胞和OTUD7A敲除的HAP1细胞系中,蛋白酶体复合物的形成和功能显着降低。我们提供证据,双等位基因致病性OTUD7A变异与早发性癫痫性脑病和蛋白酶体功能障碍有关。










































 京公网安备 11010802027423号
京公网安备 11010802027423号