当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Resolving a puzzling anomaly in the spin‐coupled generalized valence bond description of benzene
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-03-03 , DOI: 10.1002/jcc.26185 Lu T Xu 1 , David L Cooper 2 , Thom H Dunning 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-03-03 , DOI: 10.1002/jcc.26185 Lu T Xu 1 , David L Cooper 2 , Thom H Dunning 1
Affiliation
|
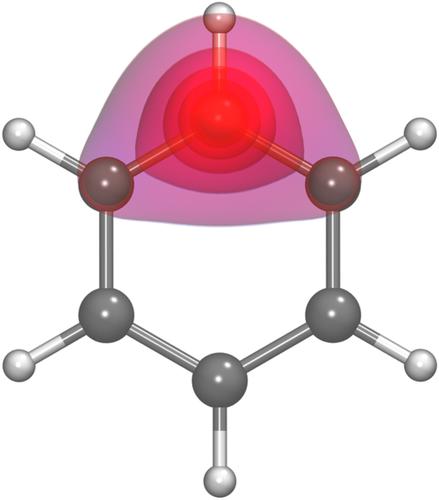
In an earlier study of benzene, Small and Head‐Gordon found that the spin‐coupled generalized valence bond (SCGVB) wave function for the π system predicted a distorted (non‐D6h) geometry, one with alternating CC bond lengths. However, the variations in the energy were very small and the predictions were made using a very small basis set (STO‐3G). We re‐examined this prediction using a much larger basis set (aug‐cc‐pVTZ) to determine the dependence of the energy of benzene on the distortion angle, ΔθCXC (ΔθCXC = 0° corresponds to the D6h structure). We also found a distorted geometry with the optimum ΔθCXC being 0.31° with an energy 0.040 kcal mol−1 lower than that for the D6h structure. In the optimum geometry, adjacent CC bond lengths are 1.3861 Å and 1.4004 Å. Analysis of the SCGVB wave function led us to conclude that the cause of the unusual non‐D6h geometry predicted by the SCGVB calculations seems to be a result of the interaction between the Kekulé and Dewar components of the full SCGVB wave function. The addition of doubly ionic configurations to the SCGVB wave function leads to the prediction of a D6h geometry for benzene and a dependence on ΔθCXC essentially the same as that predicted by the complete active space self‐consistent field wave function.
中文翻译:

解决苯的自旋耦合广义价键描述中的一个令人费解的异常
在早期对苯的研究中,Small 和 Head-Gordon 发现 π 系统的自旋耦合广义价键 (SCGVB) 波函数预测了扭曲的(非 D6h)几何形状,具有交替的 CC 键长。然而,能量的变化非常小,并且使用非常小的基组 (STO-3G) 进行预测。我们使用更大的基组 (aug-cc-pVTZ) 重新检查了这一预测,以确定苯的能量对畸变角 ΔθCXC 的依赖性(ΔθCXC = 0° 对应于 D6h 结构)。我们还发现了扭曲的几何形状,最佳 ΔθCXC 为 0.31°,能量比 D6h 结构低 0.040 kcal mol-1。在最佳几何形状中,相邻的 CC 键长为 1.3861 Å 和 1.4004 Å。对 SCGVB 波函数的分析使我们得出结论,SCGVB 计算预测的异常非 D6h 几何形状的原因似乎是完整 SCGVB 波函数的凯库勒和杜瓦组件之间相互作用的结果。在 SCGVB 波函数中添加双离子构型导致苯的 D6h 几何结构的预测以及对 ΔθCXC 的依赖与完全活性空间自洽场波函数预测的基本相同。
更新日期:2020-03-03
中文翻译:
解决苯的自旋耦合广义价键描述中的一个令人费解的异常
在早期对苯的研究中,Small 和 Head-Gordon 发现 π 系统的自旋耦合广义价键 (SCGVB) 波函数预测了扭曲的(非 D6h)几何形状,具有交替的 CC 键长。然而,能量的变化非常小,并且使用非常小的基组 (STO-3G) 进行预测。我们使用更大的基组 (aug-cc-pVTZ) 重新检查了这一预测,以确定苯的能量对畸变角 ΔθCXC 的依赖性(ΔθCXC = 0° 对应于 D6h 结构)。我们还发现了扭曲的几何形状,最佳 ΔθCXC 为 0.31°,能量比 D6h 结构低 0.040 kcal mol-1。在最佳几何形状中,相邻的 CC 键长为 1.3861 Å 和 1.4004 Å。对 SCGVB 波函数的分析使我们得出结论,SCGVB 计算预测的异常非 D6h 几何形状的原因似乎是完整 SCGVB 波函数的凯库勒和杜瓦组件之间相互作用的结果。在 SCGVB 波函数中添加双离子构型导致苯的 D6h 几何结构的预测以及对 ΔθCXC 的依赖与完全活性空间自洽场波函数预测的基本相同。










































 京公网安备 11010802027423号
京公网安备 11010802027423号