Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Integration of single nucleotide variants and whole-genome DNA methylation profiles for classification of rheumatoid arthritis cases from controls
Heredity ( IF 3.1 ) Pub Date : 2020-03-03 , DOI: 10.1038/s41437-020-0301-4 Mahmoud Amiri Roudbar 1 , Mohammad Reza Mohammadabadi 2 , Ahmad Ayatollahi Mehrgardi 2 , Rostam Abdollahi-Arpanahi 3 , Mehdi Momen 4 , Gota Morota 5 , Fernando Brito Lopes 6 , Daniel Gianola 7, 8 , Guilherme J M Rosa 7, 8
Heredity ( IF 3.1 ) Pub Date : 2020-03-03 , DOI: 10.1038/s41437-020-0301-4 Mahmoud Amiri Roudbar 1 , Mohammad Reza Mohammadabadi 2 , Ahmad Ayatollahi Mehrgardi 2 , Rostam Abdollahi-Arpanahi 3 , Mehdi Momen 4 , Gota Morota 5 , Fernando Brito Lopes 6 , Daniel Gianola 7, 8 , Guilherme J M Rosa 7, 8
Affiliation
|
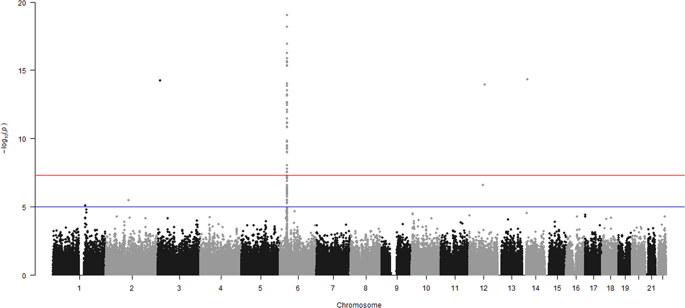
This study evaluated the use of multiomics data for classification accuracy of rheumatoid arthritis (RA). Three approaches were used and compared in terms of prediction accuracy: (1) whole-genome prediction (WGP) using SNP marker information only, (2) whole-methylome prediction (WMP) using methylation profiles only, and (3) whole-genome/methylome prediction (WGMP) with combining both omics layers. The number of SNP and of methylation sites varied in each scenario, with either 1, 10, or 50% of these preselected based on four approaches: randomly, evenly spaced, lowest p value (genome-wide association or epigenome-wide association study), and estimated effect size using a Bayesian ridge regression (BRR) model. To remove effects of high levels of pairwise linkage disequilibrium (LD), SNPs were also preselected with an LD-pruning method. Five Bayesian regression models were studied for classification, including BRR, Bayes-A, Bayes-B, Bayes-C, and the Bayesian LASSO. Adjusting methylation profiles for cellular heterogeneity within whole blood samples had a detrimental effect on the classification ability of the models. Overall, WGMP using Bayes-B model has the best performance. In particular, selecting SNPs based on LD-pruning with 1% of the methylation sites selected based on BRR included in the model, and fitting the most significant SNP as a fixed effect was the best method for predicting disease risk with a classification accuracy of 0.975. Our results showed that multiomics data can be used to effectively predict the risk of RA and identify cases in early stages to prevent or alter disease progression via appropriate interventions.
中文翻译:

整合单核苷酸变异和全基因组 DNA 甲基化图谱,用于从对照中对类风湿性关节炎病例进行分类
本研究评估了多组学数据对类风湿性关节炎 (RA) 分类准确性的使用。在预测准确性方面使用了三种方法并进行了比较:(1) 仅使用 SNP 标记信息的全基因组预测 (WGP),(2) 仅使用甲基化谱的全甲基化组预测 (WMP),以及 (3) 全基因组预测/ 甲基化组预测 (WGMP) 结合两个组学层。SNP 和甲基化位点的数量在每种情况下都不同,其中 1%、10% 或 50% 基于四种方法预选:随机、均匀间隔、最低 p 值(全基因组关联或全表观基因组关联研究) ,并使用贝叶斯岭回归 (BRR) 模型估计效果大小。为了消除高水平成对连锁不平衡 (LD) 的影响,还使用 LD 修剪方法预选了 SNP。研究了五种贝叶斯回归模型进行分类,包括 BRR、贝叶斯-A、贝叶斯-B、贝叶斯-C 和贝叶斯 LASSO。调整全血样本中细胞异质性的甲基化谱对模型的分类能力产生不利影响。总体而言,使用 Bayes-B 模型的 WGMP 具有最佳性能。尤其是基于LD剪枝选择SNP,根据模型中包含的BRR选择1%的甲基化位点,并将最显着的SNP拟合为固定效应,是预测疾病风险的最佳方法,分类准确率为0.975 . 我们的结果表明,多组学数据可用于有效预测 RA 风险并在早期识别病例,以通过适当的干预措施预防或改变疾病进展。
更新日期:2020-03-03
中文翻译:
整合单核苷酸变异和全基因组 DNA 甲基化图谱,用于从对照中对类风湿性关节炎病例进行分类
本研究评估了多组学数据对类风湿性关节炎 (RA) 分类准确性的使用。在预测准确性方面使用了三种方法并进行了比较:(1) 仅使用 SNP 标记信息的全基因组预测 (WGP),(2) 仅使用甲基化谱的全甲基化组预测 (WMP),以及 (3) 全基因组预测/ 甲基化组预测 (WGMP) 结合两个组学层。SNP 和甲基化位点的数量在每种情况下都不同,其中 1%、10% 或 50% 基于四种方法预选:随机、均匀间隔、最低 p 值(全基因组关联或全表观基因组关联研究) ,并使用贝叶斯岭回归 (BRR) 模型估计效果大小。为了消除高水平成对连锁不平衡 (LD) 的影响,还使用 LD 修剪方法预选了 SNP。研究了五种贝叶斯回归模型进行分类,包括 BRR、贝叶斯-A、贝叶斯-B、贝叶斯-C 和贝叶斯 LASSO。调整全血样本中细胞异质性的甲基化谱对模型的分类能力产生不利影响。总体而言,使用 Bayes-B 模型的 WGMP 具有最佳性能。尤其是基于LD剪枝选择SNP,根据模型中包含的BRR选择1%的甲基化位点,并将最显着的SNP拟合为固定效应,是预测疾病风险的最佳方法,分类准确率为0.975 . 我们的结果表明,多组学数据可用于有效预测 RA 风险并在早期识别病例,以通过适当的干预措施预防或改变疾病进展。










































 京公网安备 11010802027423号
京公网安备 11010802027423号