当前位置:
X-MOL 学术
›
Genet. Med.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance.
Genetics in Medicine ( IF 6.6 ) Pub Date : 2020-03-03 , DOI: 10.1038/s41436-020-0766-9 Htoo A Wai 1 , Jenny Lord 1 , Matthew Lyon 2 , Adam Gunning 3 , Hugh Kelly 1 , Penelope Cibin 1 , Eleanor G Seaby 1, 4 , Kerry Spiers-Fitzgerald 1 , Jed Lye 1 , Sian Ellard 3 , N Simon Thomas 1, 2 , David J Bunyan 1, 2 , Andrew G L Douglas 1, 5 , Diana Baralle 1, 5 ,
Genetics in Medicine ( IF 6.6 ) Pub Date : 2020-03-03 , DOI: 10.1038/s41436-020-0766-9 Htoo A Wai 1 , Jenny Lord 1 , Matthew Lyon 2 , Adam Gunning 3 , Hugh Kelly 1 , Penelope Cibin 1 , Eleanor G Seaby 1, 4 , Kerry Spiers-Fitzgerald 1 , Jed Lye 1 , Sian Ellard 3 , N Simon Thomas 1, 2 , David J Bunyan 1, 2 , Andrew G L Douglas 1, 5 , Diana Baralle 1, 5 ,
Affiliation
|
PURPOSE
Diagnosis of genetic disorders is hampered by large numbers of variants of uncertain significance (VUSs) identified through next-generation sequencing. Many such variants may disrupt normal RNA splicing. We examined effects on splicing of a large cohort of clinically identified variants and compared performance of bioinformatic splicing prediction tools commonly used in diagnostic laboratories.
METHODS
Two hundred fifty-seven variants (coding and noncoding) were referred for analysis across three laboratories. Blood RNA samples underwent targeted reverse transcription polymerase chain reaction (RT-PCR) analysis with Sanger sequencing of PCR products and agarose gel electrophoresis. Seventeen samples also underwent transcriptome-wide RNA sequencing with targeted splicing analysis based on Sashimi plot visualization. Bioinformatic splicing predictions were obtained using Alamut, HSF 3.1, and SpliceAI software.
RESULTS
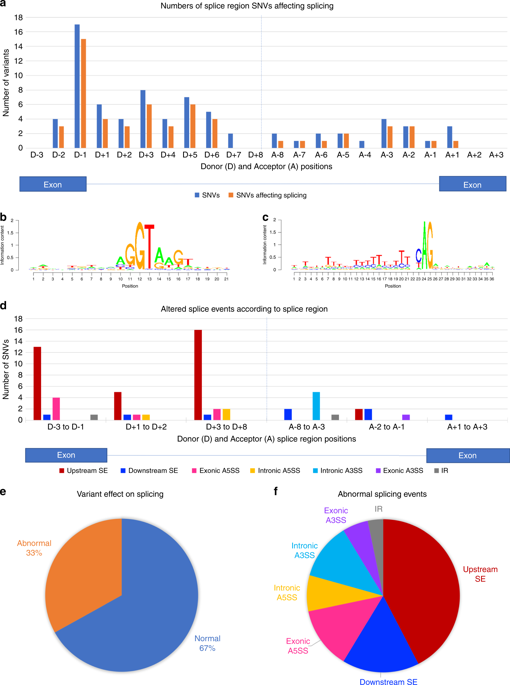
Eighty-five variants (33%) were associated with abnormal splicing. The most frequent abnormality was upstream exon skipping (39/85 variants), which was most often associated with splice donor region variants. SpliceAI had greatest accuracy in predicting splicing abnormalities (0.91) and outperformed other tools in sensitivity and specificity.
CONCLUSION
Splicing analysis of blood RNA identifies diagnostically important splicing abnormalities and clarifies functional effects of a significant proportion of VUSs. Bioinformatic predictions are improving but still make significant errors. RNA analysis should therefore be routinely considered in genetic disease diagnostics.
中文翻译:

血液 RNA 分析可以提高临床诊断率并解决不确定意义的变异。
目的 遗传疾病的诊断受到通过下一代测序鉴定的大量不确定意义的变体 (VUS) 的阻碍。许多这样的变体可能会破坏正常的 RNA 剪接。我们检查了一大群临床鉴定的变体对剪接的影响,并比较了诊断实验室中常用的生物信息学剪接预测工具的性能。方法 257 个变体(编码和非编码)被转介到三个实验室进行分析。血液 RNA 样本通过 PCR 产物的 Sanger 测序和琼脂糖凝胶电泳进行靶向逆转录聚合酶链反应 (RT-PCR) 分析。17 个样本还进行了全转录组 RNA 测序,并基于 Sashimi 图可视化进行了靶向剪接分析。使用 Alamut、HSF 3.1 和 SpliceAI 软件获得生物信息学剪接预测。结果 85 个变异 (33%) 与异常剪接相关。最常见的异常是上游外显子跳跃(39/85 变体),这通常与剪接供体区域变体有关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。结果 85 个变异 (33%) 与异常剪接相关。最常见的异常是上游外显子跳跃(39/85 变体),这通常与剪接供体区域变体有关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。结果 85 个变异 (33%) 与异常剪接相关。最常见的异常是上游外显子跳跃(39/85 变体),这通常与剪接供体区域变体有关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。这通常与剪接供体区域变体相关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。这通常与剪接供体区域变体相关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。
更新日期:2020-04-24
中文翻译:
血液 RNA 分析可以提高临床诊断率并解决不确定意义的变异。
目的 遗传疾病的诊断受到通过下一代测序鉴定的大量不确定意义的变体 (VUS) 的阻碍。许多这样的变体可能会破坏正常的 RNA 剪接。我们检查了一大群临床鉴定的变体对剪接的影响,并比较了诊断实验室中常用的生物信息学剪接预测工具的性能。方法 257 个变体(编码和非编码)被转介到三个实验室进行分析。血液 RNA 样本通过 PCR 产物的 Sanger 测序和琼脂糖凝胶电泳进行靶向逆转录聚合酶链反应 (RT-PCR) 分析。17 个样本还进行了全转录组 RNA 测序,并基于 Sashimi 图可视化进行了靶向剪接分析。使用 Alamut、HSF 3.1 和 SpliceAI 软件获得生物信息学剪接预测。结果 85 个变异 (33%) 与异常剪接相关。最常见的异常是上游外显子跳跃(39/85 变体),这通常与剪接供体区域变体有关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。结果 85 个变异 (33%) 与异常剪接相关。最常见的异常是上游外显子跳跃(39/85 变体),这通常与剪接供体区域变体有关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。结果 85 个变异 (33%) 与异常剪接相关。最常见的异常是上游外显子跳跃(39/85 变体),这通常与剪接供体区域变体有关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。这通常与剪接供体区域变体相关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。这通常与剪接供体区域变体相关。SpliceAI 在预测剪接异常方面具有最高的准确性 (0.91),并且在敏感性和特异性方面优于其他工具。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。结论 血液 RNA 的剪接分析确定了诊断上重要的剪接异常,并阐明了很大一部分 VUS 的功能影响。生物信息学预测正在改进,但仍然会出现重大错误。因此,在遗传疾病诊断中应常规考虑 RNA 分析。






































 京公网安备 11010802027423号
京公网安备 11010802027423号