Nature Methods ( IF 36.1 ) Pub Date : 2020-03-02 , DOI: 10.1038/s41592-020-0748-5 Gregory W Schwartz 1, 2 , Yeqiao Zhou 1, 2 , Jelena Petrovic 1, 2 , Maria Fasolino 3, 4 , Lanwei Xu 1, 2 , Sydney M Shaffer 1, 2 , Warren S Pear 1, 2 , Golnaz Vahedi 3, 4 , Robert B Faryabi 1, 2
|
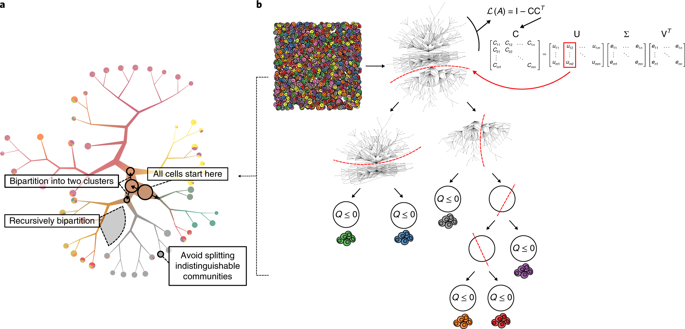
Identifying and visualizing transcriptionally similar cells is instrumental for accurate exploration of the cellular diversity revealed by single-cell transcriptomics. However, widely used clustering and visualization algorithms produce a fixed number of cell clusters. A fixed clustering ‘resolution’ hampers our ability to identify and visualize echelons of cell states. We developed TooManyCells, a suite of graph-based algorithms for efficient and unbiased identification and visualization of cell clades. TooManyCells introduces a visualization model built on a concept intentionally orthogonal to dimensionality-reduction methods. TooManyCells is also equipped with an efficient matrix-free divisive hierarchical spectral clustering different from prevalent single-resolution clustering methods. TooManyCells enables multiresolution and multifaceted exploration of single-cell clades. An advantage of this paradigm is the immediate detection of rare and common populations that outperforms popular clustering and visualization algorithms, as demonstrated using existing single-cell transcriptomic data sets and new data modeling drug-resistance acquisition in leukemic T cells.
中文翻译:
TooManyCells 识别并可视化单细胞进化枝的关系
识别和可视化转录相似的细胞有助于准确探索单细胞转录组学揭示的细胞多样性。然而,广泛使用的聚类和可视化算法会产生固定数量的细胞簇。固定的聚类“分辨率”阻碍了我们识别和可视化细胞状态梯队的能力。我们开发了 TooManyCells,这是一套基于图形的算法,用于高效、公正地识别和可视化细胞进化枝。 TooManyCells 引入了一种可视化模型,该模型建立在有意与降维方法正交的概念之上。 TooManyCells 还配备了与流行的单分辨率聚类方法不同的高效无矩阵分裂分层谱聚类。 TooManyCells 能够对单细胞进化枝进行多分辨率和多方面的探索。这种范例的一个优点是可以立即检测稀有和常见群体,其性能优于流行的聚类和可视化算法,正如使用现有的单细胞转录组数据集和白血病 T 细胞中耐药性采集的新数据建模所证明的那样。










































 京公网安备 11010802027423号
京公网安备 11010802027423号