当前位置:
X-MOL 学术
›
Mol. Phylogenet. Evol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Reconstructing phylogenetic relationships based on repeat sequence similarities.
Molecular Phylogenetics and Evolution ( IF 3.6 ) Pub Date : 2020-02-28 , DOI: 10.1016/j.ympev.2020.106766 Daniel Vitales 1 , Sònia Garcia 2 , Steven Dodsworth 3
Molecular Phylogenetics and Evolution ( IF 3.6 ) Pub Date : 2020-02-28 , DOI: 10.1016/j.ympev.2020.106766 Daniel Vitales 1 , Sònia Garcia 2 , Steven Dodsworth 3
Affiliation
|
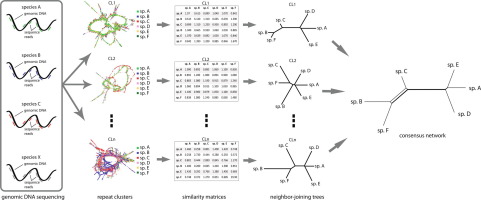
A recent phylogenetic method based on genome-wide abundance of different repeat types proved to be useful in reconstructing the evolutionary history of several plant and animal groups. Here, we demonstrate that an alternative information source from the repeatome can also be employed to infer phylogenetic relationships among taxa. Specifically, this novel approach makes use of the repeat sequence similarity matrices obtained from the comparative clustering analyses of RepeatExplorer 2, which are subsequently transformed to between-taxa distance matrices. These pairwise matrices are used to construct neighbour-joining trees for each of the top most-abundant clusters and they are finally summarized in a consensus network. This methodology was tested on three groups of angiosperms and one group of insects, resulting in congruent evolutionary hypotheses compared to more standard systematic analyses based on commonly used DNA markers. We propose that the combined application of these phylogenetic approaches based on repeat abundances and repeat sequence similarities could be helpful to understand mechanisms governing genome and repeatome evolution.
中文翻译:

基于重复序列相似性重建系统发育关系。
一种基于不同重复类型的全基因组丰度的最新系统进化方法被证明可用于重建几个动植物群体的进化史。在这里,我们证明了来自重复组的替代信息源也可以用于推断类群之间的系统发生关系。具体地说,这种新颖的方法利用了从RepeatExplorer 2的比较聚类分析中获得的重复序列相似性矩阵,这些矩阵随后被转换为类间距离矩阵。这些成对矩阵用于为每个最丰富的集群构建邻居连接树,最后将它们汇总在共识网络中。在三组被子植物和一组昆虫上测试了这种方法,与基于常用DNA标记的更标准的系统分析相比,可以得出一致的进化假设。我们建议,基于重复丰度和重复序列相似性的这些系统发育方法的组合应用可能有助于理解控制基因组和重复基因组进化的机制。
更新日期:2020-03-02
中文翻译:
基于重复序列相似性重建系统发育关系。
一种基于不同重复类型的全基因组丰度的最新系统进化方法被证明可用于重建几个动植物群体的进化史。在这里,我们证明了来自重复组的替代信息源也可以用于推断类群之间的系统发生关系。具体地说,这种新颖的方法利用了从RepeatExplorer 2的比较聚类分析中获得的重复序列相似性矩阵,这些矩阵随后被转换为类间距离矩阵。这些成对矩阵用于为每个最丰富的集群构建邻居连接树,最后将它们汇总在共识网络中。在三组被子植物和一组昆虫上测试了这种方法,与基于常用DNA标记的更标准的系统分析相比,可以得出一致的进化假设。我们建议,基于重复丰度和重复序列相似性的这些系统发育方法的组合应用可能有助于理解控制基因组和重复基因组进化的机制。








































 京公网安备 11010802027423号
京公网安备 11010802027423号