European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2020-02-28 , DOI: 10.1016/j.ejmech.2020.112195 Rachel M. Wypych , Steven R. LaPlante , Peter W. White , Stephen F. Martin
|
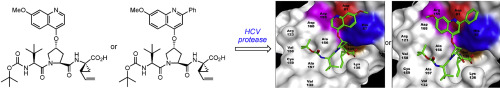
Thermodynamic parameters were determined for structurally-related inhibitors of HCV NS3 protease to assess how binding entropies and enthalpies vary with incremental changes at the P2 and P3 inhibitor subsites. Changing the heterocyclic substituent at P2 from a pyridyl to a 7-methoxy-2-phenyl-4-quinolyl group leads to a 710-fold increase in affinity. Annelating a benzene ring onto a pyridine ring leads to quinoline-derived inhibitors having higher affinities, but the individual enthalpy and entropy contributions are markedly different for each ligand pair. Introducing a phenyl group at C2 of the heterocyclic ring at P2 uniformly leads to higher affinity analogs with more favorable binding entropies, while adding a methoxy group at C7 of the quinoline ring at P2 provides derivatives with more favorable binding enthalpies. Significant enthalpy/entropy compensation is observed for structural changes made to inhibitors lacking a 2-phenyl substituent, whereas favorable changes in both binding enthalpies and entropies accompany structural modifications when a 2-phenyl group is present. Overall, binding energetics of inhibitors having a 2-phenyl-4-quinolyl group at P2 are dominated by entropic effects, whereas binding of the corresponding norphenyl analogs are primarily enthalpy driven. Notably, the reversal from an entropy driven association to an enthalpy driven one for this set of inhibitors also correlates with alternate binding modes. When the steric bulk of the side chain at P3 is increased from a hydrogen atom to a tert-butyl group, there is a 770-fold improvement in affinity. The 30-fold increase resulting from the first methyl group is solely the consequence of a more favorable change in entropy, whereas subsequent additions of methyl groups leads to modest increases in affinity that arise primarily from incremental improvements in binding enthalpies accompanied with smaller favorable entropic contributions.
中文翻译:
丙型肝炎病毒NS3蛋白酶抑制剂的结构-热力学关系
确定了HCV NS3蛋白酶的结构相关抑制剂的热力学参数,以评估结合熵和焓如何随P2和P3抑制剂亚位点的增量变化而变化。将P2处的杂环取代基从吡啶基更改为7-甲氧基-2-苯基-4-喹啉基可导致亲和力增加710倍。将苯环退火到吡啶环上会导致喹啉衍生的抑制剂具有更高的亲和力,但每个配体对的焓和熵贡献均明显不同。在P2的杂环的C2处引入苯基均匀地导致具有更有利的结合熵的较高亲和力类似物,而在P2的喹啉环的C7处加入甲氧基提供具有更有利的结合焓的衍生物。对于缺少2-苯基取代基的抑制剂的结构变化观察到显着的焓/熵补偿,而当存在2-苯基基团时,结合焓和熵的有利变化都伴随着结构修饰。总的来说,在P2处具有2-苯基-4-喹啉基的抑制剂的结合能受熵作用支配,而相应的去甲苯基类似物的结合主要是由焓驱动的。值得注意的是,对于这组抑制剂,从熵驱动的缔合转变为焓驱动的缔合也与替代的结合模式相关。当P3上侧链的空间体积从氢原子增加到 当存在2-苯基时,结合焓和熵的有利变化都伴随着结构修饰。总的来说,在P2处具有2-苯基-4-喹啉基的抑制剂的结合能受熵作用支配,而相应的去甲苯基类似物的结合主要是由焓驱动的。值得注意的是,对于这组抑制剂,从熵驱动的缔合转变为焓驱动的缔合也与替代的结合模式相关。当P3上侧链的空间体积从氢原子增加到 当存在2-苯基时,结合焓和熵的有利变化都伴随着结构修饰。总的来说,在P2处具有2-苯基-4-喹啉基的抑制剂的结合能受熵作用支配,而相应的去甲苯基类似物的结合主要是由焓驱动的。值得注意的是,对于这组抑制剂,从熵驱动的缔合转变为焓驱动的缔合也与替代的结合模式相关。当P3上侧链的空间体积从氢原子增加到 而相应的去甲苯基类似物的结合主要是由焓驱动的。值得注意的是,对于这组抑制剂,从熵驱动的缔合转变为焓驱动的缔合也与替代的结合模式相关。当P3上侧链的空间体积从氢原子增加到 而相应的去甲苯基类似物的结合主要是由焓驱动的。值得注意的是,对于这组抑制剂,从熵驱动的缔合转变为焓驱动的缔合也与替代的结合模式相关。当P3上侧链的空间体积从氢原子增加到叔丁基的亲和力提高了770倍。第一个甲基基团导致的30倍增加仅是熵的更有利变化的结果,而随后添加甲基基团则导致亲和力适度增加,这主要是由于结合焓的增量提高以及较小的有利熵贡献引起的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号