当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational screening of transition metal/p-block hybrid electrocatalysts for CO2 reduction
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-02-26 , DOI: 10.1002/jcc.26182 Sahithi Ananthaneni 1 , Rees B Rankin 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-02-26 , DOI: 10.1002/jcc.26182 Sahithi Ananthaneni 1 , Rees B Rankin 1
Affiliation
|
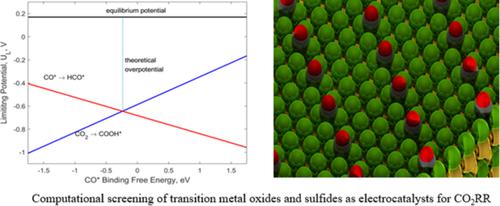
Among all the pollutants in the atmosphere, CO2 has the highest impact on global warming, and with the rising levels of this pollutant, studies on developing various technologies to convert CO2 into carbon‐neutral fuels and chemicals have become more valuable. In this work, we present a detailed computational study of electrochemical reduction of CO2 reaction (the CO2RR) to methane and/or methanol over different transition metal‐p block catalysts using density functional theory calculations. In addition to the catalyst structure, we studied reaction mechanisms using free energy diagrams that explain the product selectivity with respect to the competing hydrogen evolution reaction. Furthermore, we developed scaling relations between all the active C bound intermediate species with ΔG(CO*) and O bound species with ΔG(OH*) The limiting potential lines with ΔG(OH*) as the descriptor are much less negative compared to UL lines with ΔG(CO*) as the descriptor indicating that catalyst materials following pathways via OH− bound intermediate species require more negative potentials than CO*→ HCO* and CO2→ COOH* steps to convert into products. We developed thermodynamic volcano plots with two descriptors; the CO* and OH* binding free energies and determined the best catalyst material among the initially investigated catalyst materials expecting this plot will provide guidance to the future work on improving the activity of transition metal‐p block catalysts for this important reduction reaction.
中文翻译:

用于 CO2 还原的过渡金属/p 嵌段混合电催化剂的计算筛选
在大气中的所有污染物中,CO2 对全球变暖的影响最大,随着该污染物水平的升高,开发各种将 CO2 转化为碳中性燃料和化学品的技术的研究变得更加有价值。在这项工作中,我们使用密度泛函理论计算在不同的过渡金属-p 嵌段催化剂上对 CO2 反应(CO2RR)电化学还原为甲烷和/或甲醇进行了详细的计算研究。除了催化剂结构之外,我们还使用自由能图研究了反应机制,这些图解释了与竞争性析氢反应相关的产物选择性。此外,我们开发了具有 ΔG(CO*) 的所有活性 C 结合中间物种和具有 ΔG(OH*) 的 O 结合物种之间的比例关系 与 UL 线相比,以 ΔG(OH*) 作为描述符的极限电位线的负性要小得多ΔG(CO*) 作为描述符,表明催化剂材料通过 OH− 结合的中间物种路径需要比 CO*→HCO* 和 CO2→COOH* 步骤更多的负电位才能转化为产物。我们开发了具有两个描述符的热力学火山图;CO* 和 OH* 结合自由能,并确定了最初研究的催化剂材料中最好的催化剂材料,预计该图将为未来提高过渡金属-p 嵌段催化剂对这一重要还原反应的活性提供指导。
更新日期:2020-02-26
中文翻译:
用于 CO2 还原的过渡金属/p 嵌段混合电催化剂的计算筛选
在大气中的所有污染物中,CO2 对全球变暖的影响最大,随着该污染物水平的升高,开发各种将 CO2 转化为碳中性燃料和化学品的技术的研究变得更加有价值。在这项工作中,我们使用密度泛函理论计算在不同的过渡金属-p 嵌段催化剂上对 CO2 反应(CO2RR)电化学还原为甲烷和/或甲醇进行了详细的计算研究。除了催化剂结构之外,我们还使用自由能图研究了反应机制,这些图解释了与竞争性析氢反应相关的产物选择性。此外,我们开发了具有 ΔG(CO*) 的所有活性 C 结合中间物种和具有 ΔG(OH*) 的 O 结合物种之间的比例关系 与 UL 线相比,以 ΔG(OH*) 作为描述符的极限电位线的负性要小得多ΔG(CO*) 作为描述符,表明催化剂材料通过 OH− 结合的中间物种路径需要比 CO*→HCO* 和 CO2→COOH* 步骤更多的负电位才能转化为产物。我们开发了具有两个描述符的热力学火山图;CO* 和 OH* 结合自由能,并确定了最初研究的催化剂材料中最好的催化剂材料,预计该图将为未来提高过渡金属-p 嵌段催化剂对这一重要还原反应的活性提供指导。










































 京公网安备 11010802027423号
京公网安备 11010802027423号