当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Origin of pressure‐induced insulator‐to‐metal transition in the van der Waals compound FePS 3 from first‐principles calculations
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-02-24 , DOI: 10.1002/jcc.26178 Robert A Evarestov 1 , Alexei Kuzmin 2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-02-24 , DOI: 10.1002/jcc.26178 Robert A Evarestov 1 , Alexei Kuzmin 2
Affiliation
|
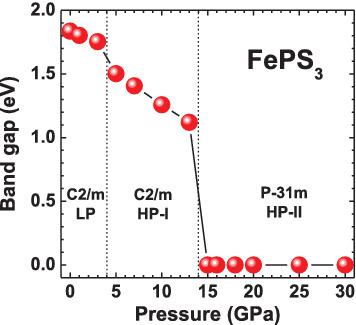
Pressure‐induced insulator‐to‐metal transition (IMT) has been studied in the van der Waals compound iron thiophosphate (FePS3) using first‐principles calculations within the periodic linear combination of atomic orbitals method with hybrid Hartree–Fock‐DFT B3LYP functional. Our calculations reproduce correctly the IMT at ∼15 GPa, which is accompanied by a reduction of the unit cell volume and of the vdW gap. We found from the detailed analysis of the projected density of states that the 3p states of phosphorus atoms contribute significantly at the bottom of the conduction band. As a result, the collapse of the band gap occurs due to changes in the electronic structure of FePS3 induced by relative displacements of phosphorus or sulfur atoms along the c‐axis direction under pressure.
中文翻译:

第一性原理计算范德华化合物FePS 3 中压力诱导绝缘体到金属转变的起源
已经在范德华化合物硫代磷酸铁(FePS3)中使用第一性原理计算在原子轨道方法与混合 Hartree-Fock-DFT B3LYP 函数的周期性线性组合中研究了压力诱导绝缘体到金属的转变(IMT)。我们的计算正确地再现了约 15 GPa 的 IMT,伴随着晶胞体积和 vdW 间隙的减小。我们从投影态密度的详细分析中发现,磷原子的 3p 态在导带底部有显着贡献。结果,由于磷或硫原子在压力下沿 c 轴方向的相对位移引起的 FePS3 电子结构的变化,导致带隙坍塌。
更新日期:2020-02-24
中文翻译:
第一性原理计算范德华化合物FePS 3 中压力诱导绝缘体到金属转变的起源
已经在范德华化合物硫代磷酸铁(FePS3)中使用第一性原理计算在原子轨道方法与混合 Hartree-Fock-DFT B3LYP 函数的周期性线性组合中研究了压力诱导绝缘体到金属的转变(IMT)。我们的计算正确地再现了约 15 GPa 的 IMT,伴随着晶胞体积和 vdW 间隙的减小。我们从投影态密度的详细分析中发现,磷原子的 3p 态在导带底部有显着贡献。结果,由于磷或硫原子在压力下沿 c 轴方向的相对位移引起的 FePS3 电子结构的变化,导致带隙坍塌。










































 京公网安备 11010802027423号
京公网安备 11010802027423号