当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Development of Density Functional Tight-Binding Parameters Using Relative Energy Fitting and Particle Swarm Optimization.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-02-20 , DOI: 10.1021/acs.jctc.9b00880 Néstor F Aguirre 1 , Amanda Morgenstern 1 , M J Cawkwell 1 , Enrique R Batista 1 , Ping Yang 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-02-20 , DOI: 10.1021/acs.jctc.9b00880 Néstor F Aguirre 1 , Amanda Morgenstern 1 , M J Cawkwell 1 , Enrique R Batista 1 , Ping Yang 1
Affiliation
|
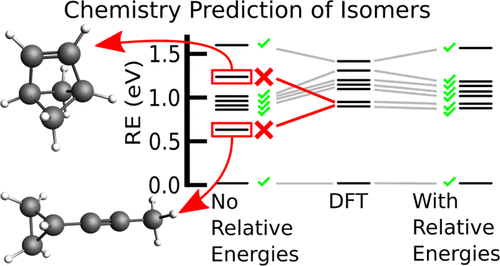
We provide a strategy to optimize density functional tight-binding (DFTB) parameterization for the calculation of the structures and properties of organic molecules consisting of hydrogen, carbon, nitrogen, and oxygen. We utilize an objective function based on similarity measurements and the Particle Swarm Optimization (PSO) method to find an optimal set of parameters. This objective function considers not only the common DFTB descriptors of binding energies and atomic forces but also incorporates relative energies of isomers into the fitting procedure for more chemistry-driven results. The quality in the description of the binding energies and atomic forces is measured based on the Ballester similarity index and relative energies through a similarity index induced by the Levenshtein edit distance to quantify the correct energetic order of isomers. Training and testing datasets were created to include all relevant chemical functional groups. The accuracy of this strategy is assessed, and its range of applicability is discussed by comparison against our previous parameterization [A. Krishnapriyan, et al., J. Chem. Theory Comput. 13, 6191 (2017)]. The improved performance of the new DFTB parameterization is validated with respect to the density functional theory large datasets QM-9 [R. Ramakrishnan, et al., Sci. Data 1, 140022 (2014)] and ANI-1 [J. S. Smith, et al., Sci. Data 4, 170193 (2017)], where excellent agreement is found between the structures and properties available in these datasets, and the ones obtained with DFTB.
中文翻译:

利用相对能量拟合和粒子群算法开发密度泛函紧束缚参数。
我们提供了一种优化密度泛函紧密结合(DFTB)参数化的策略,用于计算由氢,碳,氮和氧组成的有机分子的结构和性质。我们利用基于相似性度量的目标函数和粒子群优化(PSO)方法来找到最佳参数集。该目标函数不仅考虑结合能和原子力的通用DFTB描述子,而且还将异构体的相对能纳入拟合过程中,以得到更多化学驱动的结果。结合能和原子力的描述中的质量是基于巴列斯特相似指数和相对能量通过莱文施泰因编辑距离诱导的相似指数来测量的,以量化异构体的正确能级。创建了培训和测试数据集,以包括所有相关的化学官能团。评估了该策略的准确性,并通过与我们之前的参数化比较来讨论了其适用范围[A. 克里希纳普里扬(Krishnapriyan)等人,化学杂志(J. Chem。)理论计算。13,6191(2017)]。相对于密度泛函理论大型数据集QM-9 [R。Ramakrishnan等,科学。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。通过与我们先前的参数化比较来讨论其适用范围。克里希纳普里扬(Krishnapriyan)等人,化学杂志(J. Chem。)理论计算。13,6191(2017)]。相对于密度泛函理论大型数据集QM-9 [R.],验证了新DFTB参数化的改进性能。Ramakrishnan等,科学。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。通过与我们先前的参数化比较来讨论其适用范围。克里希纳普里扬(Krishnapriyan)等人,化学杂志(J. Chem。)理论计算。13,6191(2017)]。相对于密度泛函理论大型数据集QM-9 [R。Ramakrishnan等,科学。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。
更新日期:2020-02-21
中文翻译:
利用相对能量拟合和粒子群算法开发密度泛函紧束缚参数。
我们提供了一种优化密度泛函紧密结合(DFTB)参数化的策略,用于计算由氢,碳,氮和氧组成的有机分子的结构和性质。我们利用基于相似性度量的目标函数和粒子群优化(PSO)方法来找到最佳参数集。该目标函数不仅考虑结合能和原子力的通用DFTB描述子,而且还将异构体的相对能纳入拟合过程中,以得到更多化学驱动的结果。结合能和原子力的描述中的质量是基于巴列斯特相似指数和相对能量通过莱文施泰因编辑距离诱导的相似指数来测量的,以量化异构体的正确能级。创建了培训和测试数据集,以包括所有相关的化学官能团。评估了该策略的准确性,并通过与我们之前的参数化比较来讨论了其适用范围[A. 克里希纳普里扬(Krishnapriyan)等人,化学杂志(J. Chem。)理论计算。13,6191(2017)]。相对于密度泛函理论大型数据集QM-9 [R。Ramakrishnan等,科学。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。通过与我们先前的参数化比较来讨论其适用范围。克里希纳普里扬(Krishnapriyan)等人,化学杂志(J. Chem。)理论计算。13,6191(2017)]。相对于密度泛函理论大型数据集QM-9 [R.],验证了新DFTB参数化的改进性能。Ramakrishnan等,科学。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。通过与我们先前的参数化比较来讨论其适用范围。克里希纳普里扬(Krishnapriyan)等人,化学杂志(J. Chem。)理论计算。13,6191(2017)]。相对于密度泛函理论大型数据集QM-9 [R。Ramakrishnan等,科学。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。数据1,140022(2014)]和ANI-1 [JS Smith等,Sci。数据4,170193(2017)],其中在这些数据集中可用的结构和属性与使用DFTB获得的结构和属性之间找到了极好的一致性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号