Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
DFT/TD-DFT investigation on the photoinduced electron transfer of diruthenium and viologen complexes
Journal of Luminescence ( IF 3.3 ) Pub Date : 2020-06-01 , DOI: 10.1016/j.jlumin.2020.117121 Malinee Promkatkaew , Songwut Suramitr , Thitinun Karpkird , Masahiro Ehara , Supa Hannongbua
Journal of Luminescence ( IF 3.3 ) Pub Date : 2020-06-01 , DOI: 10.1016/j.jlumin.2020.117121 Malinee Promkatkaew , Songwut Suramitr , Thitinun Karpkird , Masahiro Ehara , Supa Hannongbua
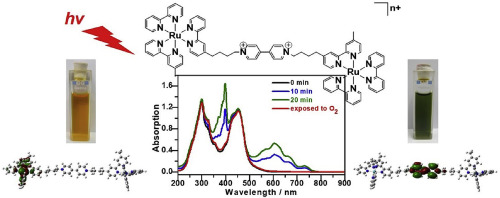
|
Abstract Photoinduced electron transfer, chemical stability, and redox property of diruthenium−viologen complexes were investigated using density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations. Target systems include a series of diruthenium and viologen complexes consisting of Ru(bpy)3-viologen-Ru(bpy)3; Ru–V–Ru, Ru(bpy)3-viologen-Ru(bpy)(dcbpy)2; Ru–V–RuCOOH, and Ru(bpy)3-viologen-viologen-Ru(bpy)3; Ru–V–V–Ru, where bpy = 2,2′-bipyridyl and dcbpy = 4,4′-dicarboxyl-2,2-bipyridyl. The B3LYP functional was used to investigate the ground state properties for the three diruthenium and viologen complexes and for two parent complexes: ruthenium-bipyridine (Ru(bpy)32+) and methyl viologen (MV2+) as well as their oxidized states. Singlet excited states were examined using the TD-DFT calculations with CAM-B3LYP functional in acetonitrile using conductor polarizable continuum model (CPCM). The TD-DFT calculations reproduced the experimentally observed variation of the absorption spectra due to oxidation states. The correlation between the calculated UV–Vis transition and molecular orbitals for the excited state clearly established that the viologen dication (V2+) changed their reductive product into the radical cation (V•+) and neutral (V0), whereas ruthenium retained its formal Ru(bpy)32+ oxidation state. The metal to ligand charge transfer state of Ru(bpy)32+ was shown to undergo electron transfer quenching by methyl viologen dication (MV2+). These findings are relevant for the development of novel optoelectronic materials owing to a combination of chemical stability, redox activity, and long-lived excited states.
中文翻译:

DFT/TD-DFT研究二钌和紫精配合物的光致电子转移
摘要 使用密度泛函理论 (DFT) 和瞬态 DFT (TD-DFT) 计算研究了二钌-紫精配合物的光致电子转移、化学稳定性和氧化还原性能。目标系统包括一系列由 Ru(bpy)3-viologen-Ru(bpy)3 组成的二钌和紫精配合物;Ru-V-Ru,Ru(bpy)3-紫精-Ru(bpy)(dcbpy)2;Ru-V-RuCOOH和Ru(bpy)3-紫精-紫精-Ru(bpy)3;Ru-V-V-Ru,其中 bpy = 2,2'-联吡啶,dcbpy = 4,4'-二羧基-2,2-联吡啶。B3LYP 泛函用于研究三种二钌和紫精配合物以及两种母体配合物的基态特性:钌-联吡啶 (Ru(bpy)32+) 和甲基紫精 (MV2+) 以及它们的氧化态。使用导体极化连续介质模型 (CPCM) 使用 CAM-B3LYP 功能在乙腈中使用 TD-DFT 计算检查单线态激发态。TD-DFT 计算再现了实验观察到的由于氧化态引起的吸收光谱的变化。计算出的 UV-Vis 跃迁与激发态分子轨道之间的相关性清楚地表明,紫精二阳离子 (V2+) 将其还原产物变为自由基阳离子 (V•+) 和中性 (V0),而钌保留其形式的 Ru (bpy)32+ 氧化态。Ru(bpy)32+ 的金属到配体电荷转移状态显示出通过甲基紫精二化 (MV2+) 进行电子转移猝灭。由于化学稳定性,这些发现与新型光电材料的开发有关,
更新日期:2020-06-01
中文翻译:
DFT/TD-DFT研究二钌和紫精配合物的光致电子转移
摘要 使用密度泛函理论 (DFT) 和瞬态 DFT (TD-DFT) 计算研究了二钌-紫精配合物的光致电子转移、化学稳定性和氧化还原性能。目标系统包括一系列由 Ru(bpy)3-viologen-Ru(bpy)3 组成的二钌和紫精配合物;Ru-V-Ru,Ru(bpy)3-紫精-Ru(bpy)(dcbpy)2;Ru-V-RuCOOH和Ru(bpy)3-紫精-紫精-Ru(bpy)3;Ru-V-V-Ru,其中 bpy = 2,2'-联吡啶,dcbpy = 4,4'-二羧基-2,2-联吡啶。B3LYP 泛函用于研究三种二钌和紫精配合物以及两种母体配合物的基态特性:钌-联吡啶 (Ru(bpy)32+) 和甲基紫精 (MV2+) 以及它们的氧化态。使用导体极化连续介质模型 (CPCM) 使用 CAM-B3LYP 功能在乙腈中使用 TD-DFT 计算检查单线态激发态。TD-DFT 计算再现了实验观察到的由于氧化态引起的吸收光谱的变化。计算出的 UV-Vis 跃迁与激发态分子轨道之间的相关性清楚地表明,紫精二阳离子 (V2+) 将其还原产物变为自由基阳离子 (V•+) 和中性 (V0),而钌保留其形式的 Ru (bpy)32+ 氧化态。Ru(bpy)32+ 的金属到配体电荷转移状态显示出通过甲基紫精二化 (MV2+) 进行电子转移猝灭。由于化学稳定性,这些发现与新型光电材料的开发有关,










































 京公网安备 11010802027423号
京公网安备 11010802027423号