Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The Methyl Torsion in Unsaturated Compounds.
ACS Omega ( IF 3.7 ) Pub Date : 2020-02-07 , DOI: 10.1021/acsomega.9b03351 Andrea Zachariou 1, 2 , Alexander P Hawkins 1, 2 , Paul Collier 3 , Russell F Howe 4 , David Lennon 1, 2 , Stewart F Parker 1, 2, 5
ACS Omega ( IF 3.7 ) Pub Date : 2020-02-07 , DOI: 10.1021/acsomega.9b03351 Andrea Zachariou 1, 2 , Alexander P Hawkins 1, 2 , Paul Collier 3 , Russell F Howe 4 , David Lennon 1, 2 , Stewart F Parker 1, 2, 5
Affiliation
|
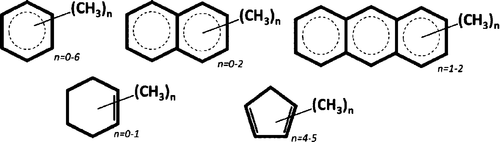
How the methyl torsion transition energy in unsaturated systems is affected by its environment is investigated. It is strongly influenced by both its immediate neighborhood, (the number of methyl groups present in the molecule) and the intermolecular interactions. It is clear that the intermolecular interactions have a major influence on the torsion transition energy, as demonstrated unambiguously previously for mesitylene and also seen here for other systems. In part, this may be caused by the fact that the methyl torsion is rarely a pure mode (unless enforced by symmetry). Where the crystal structure is available, the assignments have been supported by CASTEP calculations of the unit cell. The agreement between the observed and calculated spectra is generally good, although not perfect, toluene being a case in point, and highlights just how demanding it is to obtain accurate transition energies for low energy modes. The disagreement between observed and calculated inelastic neutron scattering spectra for meta-xylene and 9,10 dimethylanthracene is so severe that it would suggest that there are additional phases to those presently known. Comparison between the full periodic calculations and those for the isolated molecule shows that intermolecular interactions raise the methyl torsion transition energy by at least 8% and in some cases by more than 50%. The presence of more than one methyl group in the molecule generally raises the average torsion energy from the <100 cm-1 seen for single methyl groups to 150-200 cm-1.
中文翻译:

不饱和化合物中的甲基扭转。
研究了不饱和体系中甲基扭转转变能如何受到其环境的影响。它受到其直接邻域(分子中存在的甲基数量)和分子间相互作用的强烈影响。显然,分子间的相互作用对扭转跃迁能有重要影响,如以前对于均三甲苯和在其他系统中也清楚地表明的那样。在某种程度上,这可能是由于甲基扭转很少是纯模式(除非通过对称实施)的事实引起的。在晶体结构可用的情况下,单元格的CASTEP计算已支持分配。观察到的光谱与计算得出的光谱之间的一致性通常很好,尽管并不完美,甲苯就是一个很好的例子,并重点介绍了在低能耗模式下获得准确过渡能量的要求。对于间二甲苯和9,10二甲基蒽,观察到的和计算出的非弹性中子散射光谱之间的分歧如此严重,以至于暗示着目前已知的还有其他相。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基所见的<100 cm-1提高到150-200 cm-1。对于间二甲苯和9,10二甲基蒽,观察到的和计算出的非弹性中子散射光谱之间的分歧如此严重,以至于暗示着目前已知的还有其他相。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基所见的<100 cm-1提高到150-200 cm-1。对于间二甲苯和9,10二甲基蒽,观察到的和计算出的非弹性中子散射光谱之间的分歧如此严重,以至于暗示着目前已知的还有其他相。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基的<100 cm-1提高到150-200 cm-1。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基所见的<100 cm-1提高到150-200 cm-1。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基的<100 cm-1提高到150-200 cm-1。
更新日期:2020-02-18
中文翻译:
不饱和化合物中的甲基扭转。
研究了不饱和体系中甲基扭转转变能如何受到其环境的影响。它受到其直接邻域(分子中存在的甲基数量)和分子间相互作用的强烈影响。显然,分子间的相互作用对扭转跃迁能有重要影响,如以前对于均三甲苯和在其他系统中也清楚地表明的那样。在某种程度上,这可能是由于甲基扭转很少是纯模式(除非通过对称实施)的事实引起的。在晶体结构可用的情况下,单元格的CASTEP计算已支持分配。观察到的光谱与计算得出的光谱之间的一致性通常很好,尽管并不完美,甲苯就是一个很好的例子,并重点介绍了在低能耗模式下获得准确过渡能量的要求。对于间二甲苯和9,10二甲基蒽,观察到的和计算出的非弹性中子散射光谱之间的分歧如此严重,以至于暗示着目前已知的还有其他相。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基所见的<100 cm-1提高到150-200 cm-1。对于间二甲苯和9,10二甲基蒽,观察到的和计算出的非弹性中子散射光谱之间的分歧如此严重,以至于暗示着目前已知的还有其他相。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基所见的<100 cm-1提高到150-200 cm-1。对于间二甲苯和9,10二甲基蒽,观察到的和计算出的非弹性中子散射光谱之间的分歧如此严重,以至于暗示着目前已知的还有其他相。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基的<100 cm-1提高到150-200 cm-1。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基所见的<100 cm-1提高到150-200 cm-1。完整周期计算与孤立分子计算的比较表明,分子间相互作用将甲基扭转转变能提高了至少8%,在某些情况下提高了50%以上。分子中不止一个甲基的存在通常会使平均扭转能从单个甲基的<100 cm-1提高到150-200 cm-1。










































 京公网安备 11010802027423号
京公网安备 11010802027423号