当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Probing the bent bonds in cyclopropane systems for gas storage and separation process: A computational study
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-05-15 , DOI: 10.1002/jcc.26174 Padmaja D Wakchaure 1, 2 , Bishwajit Ganguly 1, 2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-05-15 , DOI: 10.1002/jcc.26174 Padmaja D Wakchaure 1, 2 , Bishwajit Ganguly 1, 2
Affiliation
|
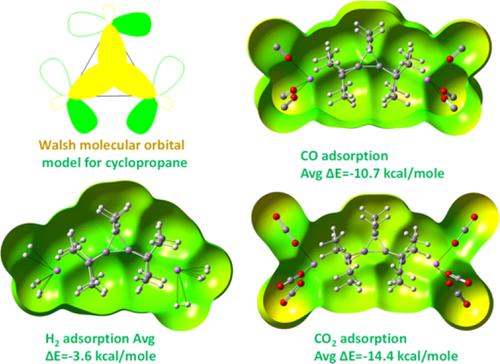
The hydrogen, carbon dioxide, and carbon monoxide gas adsorption and storage capacity of lithium‐decorated cyclopropane ring systems were examined with quantum chemical calculations at density functional theory, DFT M06‐2X functional using 6‐31G(d) and cc‐pVDZ basis sets. To examine the reliability of M06‐2X DFT functional, a few representative systems are also examined with complete basis set CBS‐QB3 method and CCSD‐aug‐cc‐pVTZ level of theory. The cyclopropane systems can bind to one Li+ ion; however, the corresponding the methylated systems can bind with two Li+ ions. The cyclopropane systems can adsorb six hydrogen molecules with an average binding energy of 3.8 kcal/mol. The binding free energy (ΔG) values suggest that the hydrogen adsorption process is feasible at 273.15 K. The calculation of desorption energies indicates the recyclable property of gas adsorbed complexes. The same number of CO2 and CO gas molecules can also be adsorbed with an average binding energy of −14.4 kcal/mol and −10.7 kcal/mol, respectively. The carbon dioxide showed ~3–4 kcal/mol better binding energy as compared to carbon monoxide and hence such designed systems can function as a potential candidate for the separation of these flue gas molecules. The nature of interactions in complexes was examined with atoms in molecules analysis revealed the electrostatic nature for the interaction of Li+ ion with cyclopropane rings. The chemical hardness and electrophilicity calculations showed that the gas adsorbed complexes are rigid and therefore robust as gas storage materials.
中文翻译:

探索用于气体储存和分离过程的环丙烷系统中的弯曲键:一项计算研究
使用 6-31G(d) 和 cc-pVDZ 基组,在密度泛函理论、DFT M06-2X 泛函、量子化学计算下,研究了锂修饰的环丙烷环系统的氢气、二氧化碳和一氧化碳气体吸附和存储能力. 为了检验 M06-2X DFT 泛函的可靠性,还使用完全基组 CBS-QB3 方法和 CCSD-aug-cc-pVTZ 理论水平检验了一些代表性系统。环丙烷系统可以结合一个锂离子;然而,相应的甲基化系统可以与两个 Li+ 离子结合。环丙烷系统可以吸附六个氢分子,平均结合能为 3.8 kcal/mol。结合自由能 (ΔG) 值表明氢吸附过程在 273.15 K 是可行的。解吸能的计算表明气体吸附复合物的可回收性。也可以吸附相同数量的 CO2 和 CO 气体分子,平均结合能分别为 -14.4 kcal/mol 和 -10.7 kcal/mol。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。也可以吸附相同数量的 CO2 和 CO 气体分子,平均结合能分别为 -14.4 kcal/mol 和 -10.7 kcal/mol。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。也可以吸附相同数量的 CO2 和 CO 气体分子,平均结合能分别为 -14.4 kcal/mol 和 -10.7 kcal/mol。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。
更新日期:2020-05-15
中文翻译:
探索用于气体储存和分离过程的环丙烷系统中的弯曲键:一项计算研究
使用 6-31G(d) 和 cc-pVDZ 基组,在密度泛函理论、DFT M06-2X 泛函、量子化学计算下,研究了锂修饰的环丙烷环系统的氢气、二氧化碳和一氧化碳气体吸附和存储能力. 为了检验 M06-2X DFT 泛函的可靠性,还使用完全基组 CBS-QB3 方法和 CCSD-aug-cc-pVTZ 理论水平检验了一些代表性系统。环丙烷系统可以结合一个锂离子;然而,相应的甲基化系统可以与两个 Li+ 离子结合。环丙烷系统可以吸附六个氢分子,平均结合能为 3.8 kcal/mol。结合自由能 (ΔG) 值表明氢吸附过程在 273.15 K 是可行的。解吸能的计算表明气体吸附复合物的可回收性。也可以吸附相同数量的 CO2 和 CO 气体分子,平均结合能分别为 -14.4 kcal/mol 和 -10.7 kcal/mol。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。也可以吸附相同数量的 CO2 和 CO 气体分子,平均结合能分别为 -14.4 kcal/mol 和 -10.7 kcal/mol。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。也可以吸附相同数量的 CO2 和 CO 气体分子,平均结合能分别为 -14.4 kcal/mol 和 -10.7 kcal/mol。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。与一氧化碳相比,二氧化碳显示出~3-4 kcal/mol 更好的结合能,因此这种设计的系统可以作为分离这些烟道气分子的潜在候选物。用分子分析中的原子检查了配合物中相互作用的性质,揭示了 Li+ 离子与环丙烷环相互作用的静电性质。化学硬度和亲电性计算表明,吸附气体的复合物是刚性的,因此作为储气材料是坚固的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号