当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Predicting whether aromatic molecules would prefer to enter a carbon nanotube: A density functional theory study
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-05-15 , DOI: 10.1002/jcc.26173 Dae-Hwan Ahn 1 , Chiyoung Park 2 , Jong-Won Song 1, 3
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-05-15 , DOI: 10.1002/jcc.26173 Dae-Hwan Ahn 1 , Chiyoung Park 2 , Jong-Won Song 1, 3
Affiliation
|
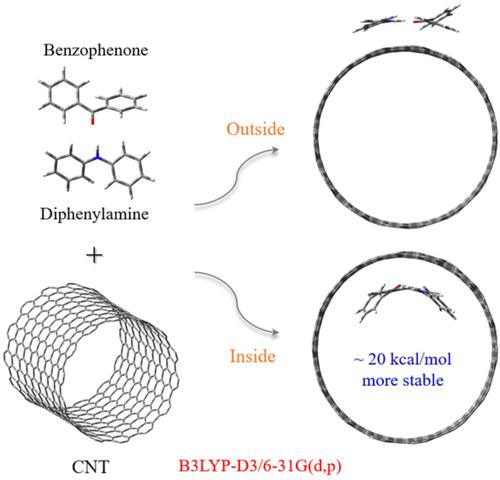
The interaction of a carbon nanotube (CNT) with various aromatic molecules, such as aniline, benzophenone, and diphenylamine, was studied using density functional theory able to compute intermolecular weak interactions (B3LYP‐D3). CNTs of varying lengths were used, such as 4‐CNT, 6‐CNT, and 8‐CNT (the numbers denoting relative lengths), with the lengths being chosen appropriately to save computation times. All aromatic molecules were found to exhibit strong intermolecular binding energies with the inner surface of the CNT, rather than the outer surface. Hydrogen bonding between two aromatic molecules that include N and O atoms is shown to further stabilize the intermolecular adsorption process. Therefore, when benzophenone and diphenylamine were simultaneously allowed to interact with a CNT, the aromatic molecules were expected to preferably enter the CNT. Furthermore, additional calculations of the intermolecular adsorption energy for aniline adsorbed on a graphene surface showed that the concavity of graphene‐like carbon sheet is in proportion to the intermolecular binding energy between the graphene‐like carbon sheet and the aromatic molecule.
中文翻译:

预测芳香分子是否更愿意进入碳纳米管:密度泛函理论研究
使用能够计算分子间弱相互作用(B3LYP-D3)的密度泛函理论研究了碳纳米管(CNT)与各种芳香族分子(如苯胺、二苯甲酮和二苯胺)的相互作用。使用不同长度的CNT,例如4-CNT,6-CNT和8-CNT(表示相对长度的数字),其长度被适当地选择以节省计算时间。发现所有芳族分子都表现出与 CNT 内表面的强分子间结合能,而不是外表面。包含 N 和 O 原子的两个芳族分子之间的氢键被证明可以进一步稳定分子间吸附过程。因此,当同时允许二苯甲酮和二苯胺与 CNT 相互作用时,预期芳香分子优选进入CNT。此外,对吸附在石墨烯表面上的苯胺的分子间吸附能的额外计算表明,类石墨烯碳片的凹度与类石墨烯碳片与芳香分子之间的分子间结合能成正比。
更新日期:2020-05-15
中文翻译:
预测芳香分子是否更愿意进入碳纳米管:密度泛函理论研究
使用能够计算分子间弱相互作用(B3LYP-D3)的密度泛函理论研究了碳纳米管(CNT)与各种芳香族分子(如苯胺、二苯甲酮和二苯胺)的相互作用。使用不同长度的CNT,例如4-CNT,6-CNT和8-CNT(表示相对长度的数字),其长度被适当地选择以节省计算时间。发现所有芳族分子都表现出与 CNT 内表面的强分子间结合能,而不是外表面。包含 N 和 O 原子的两个芳族分子之间的氢键被证明可以进一步稳定分子间吸附过程。因此,当同时允许二苯甲酮和二苯胺与 CNT 相互作用时,预期芳香分子优选进入CNT。此外,对吸附在石墨烯表面上的苯胺的分子间吸附能的额外计算表明,类石墨烯碳片的凹度与类石墨烯碳片与芳香分子之间的分子间结合能成正比。










































 京公网安备 11010802027423号
京公网安备 11010802027423号