Communications Chemistry ( IF 5.9 ) Pub Date : 2020-02-12 , DOI: 10.1038/s42004-020-0264-7 Beth A Caine 1, 2 , Maddalena Bronzato 3 , Torquil Fraser 3 , Nathan Kidley 3 , Christophe Dardonville 4 , Paul L A Popelier 1, 2
|
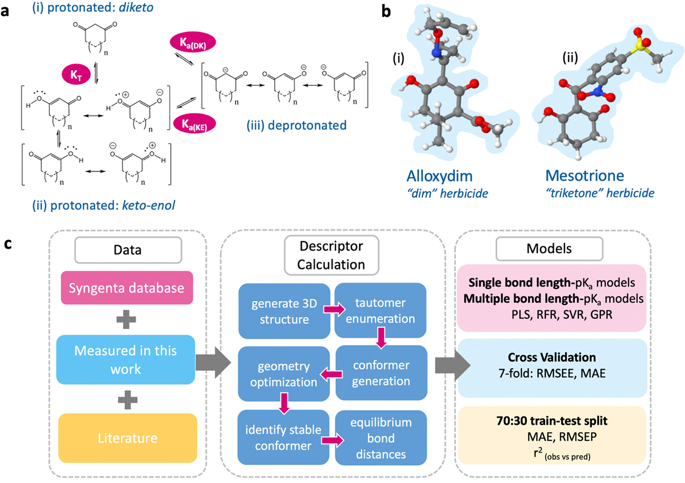
The accurate prediction of aqueous pKa values for tautomerizable compounds is a formidable task, even for the most established in silico tools. Empirical approaches often fall short due to a lack of pre-existing knowledge of dominant tautomeric forms. In a rigorous first-principles approach, calculations for low-energy tautomers must be performed in protonated and deprotonated forms, often both in gas and solvent phases, thus representing a significant computational task. Here we report an alternative approach, predicting pKa values for herbicide/therapeutic derivatives of 1,3-cyclohexanedione and 1,3-cyclopentanedione to within just 0.24 units. A model, using a single ab initio bond length from one protonation state, is as accurate as other more complex regression approaches using more input features, and outperforms the program Marvin. Our approach can be used for other tautomerizable species, to predict trends across congeneric series and to correct experimental pKa values.
中文翻译:
水性 pKa 使用平衡键长预测互变异构化合物
准确预测可互变异构化合物的水溶液 pKa值是一项艰巨的任务,即使对于最成熟的计算机工具也是如此。由于缺乏对主要互变异构形式的预先存在的知识,经验方法往往不足。在严格的第一性原理方法中,低能互变异构体的计算必须以质子化和去质子化形式进行,通常在气相和溶剂相中进行,因此是一项重要的计算任务。在这里,我们报告了一种替代方法,预测 pK a1,3-环己二酮和 1,3-环戊二酮的除草剂/治疗性衍生物的值仅在 0.24 个单位以内。使用来自一个质子化状态的单个从头算键长的模型与使用更多输入特征的其他更复杂的回归方法一样准确,并且优于程序 Marvin。我们的方法可用于其他可互变异构的物种,以预测同类系列的趋势并校正实验 pK a值。










































 京公网安备 11010802027423号
京公网安备 11010802027423号