当前位置:
X-MOL 学术
›
Chem. Rev.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Non-adiabatic Excited-State Molecular Dynamics: Theory and Applications for Modeling Photophysics in Extended Molecular Materials.
Chemical Reviews ( IF 62.1 ) Pub Date : 2020-02-10 , DOI: 10.1021/acs.chemrev.9b00447 Tammie R Nelson 1 , Alexander J White 1 , Josiah A Bjorgaard 1 , Andrew E Sifain 1, 2 , Yu Zhang 1 , Benjamin Nebgen 1 , Sebastian Fernandez-Alberti 3 , Dmitry Mozyrsky 1 , Adrian E Roitberg 4 , Sergei Tretiak 1
Chemical Reviews ( IF 62.1 ) Pub Date : 2020-02-10 , DOI: 10.1021/acs.chemrev.9b00447 Tammie R Nelson 1 , Alexander J White 1 , Josiah A Bjorgaard 1 , Andrew E Sifain 1, 2 , Yu Zhang 1 , Benjamin Nebgen 1 , Sebastian Fernandez-Alberti 3 , Dmitry Mozyrsky 1 , Adrian E Roitberg 4 , Sergei Tretiak 1
Affiliation
|
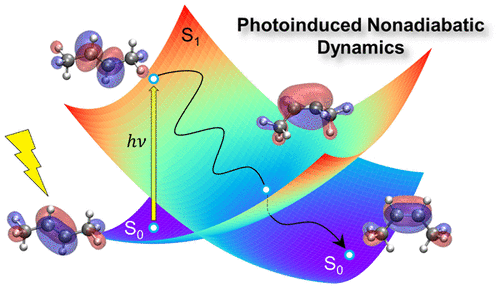
Optically active molecular materials, such as organic conjugated polymers and biological systems, are characterized by strong coupling between electronic and vibrational degrees of freedom. Typically, simulations must go beyond the Born-Oppenheimer approximation to account for non-adiabatic coupling between excited states. Indeed, non-adiabatic dynamics is commonly associated with exciton dynamics and photophysics involving charge and energy transfer, as well as exciton dissociation and charge recombination. Understanding the photoinduced dynamics in such materials is vital to providing an accurate description of exciton formation, evolution, and decay. This interdisciplinary field has matured significantly over the past decades. Formulation of new theoretical frameworks, development of more efficient and accurate computational algorithms, and evolution of high-performance computer hardware has extended these simulations to very large molecular systems with hundreds of atoms, including numerous studies of organic semiconductors and biomolecules. In this Review, we will describe recent theoretical advances including treatment of electronic decoherence in surface-hopping methods, the role of solvent effects, trivial unavoided crossings, analysis of data based on transition densities, and efficient computational implementations of these numerical methods. We also emphasize newly developed semiclassical approaches, based on the Gaussian approximation, which retain phase and width information to account for significant decoherence and interference effects while maintaining the high efficiency of surface-hopping approaches. The above developments have been employed to successfully describe photophysics in a variety of molecular materials.
中文翻译:

非绝热激发态分子动力学:扩展分子材料中光物理建模的理论和应用。
光学活性分子材料,例如有机共轭聚合物和生物系统,其特征在于电子自由度和振动自由度之间的强耦合。通常,模拟必须超出Born-Oppenheimer逼近,才能说明激发态之间的非绝热耦合。实际上,非绝热动力学通常与激子动力学和光物理相关,涉及电荷和能量转移,以及激子解离和电荷复合。了解此类材料中的光致动力学对于提供激子形成,演化和衰减的准确描述至关重要。在过去的几十年中,这个跨学科领域已经显着成熟。制定新的理论框架,开发更高效,更准确的计算算法,随着高性能计算机硬件的发展,这些模拟已扩展到具有数百个原子的超大型分子系统,包括对有机半导体和生物分子的大量研究。在这篇综述中,我们将描述最近的理论进展,包括在表面跳跃方法中处理电子去相干,溶剂效应的作用,不可避免的琐碎交叉,基于跃迁密度的数据分析以及这些数值方法的有效计算实现。我们还强调了基于高斯近似的新开发的半经典方法,该方法保留了相位和宽度信息,以解决显着的相干和干扰效应,同时保持了表面跳变方法的高效率。
更新日期:2020-02-11
中文翻译:
非绝热激发态分子动力学:扩展分子材料中光物理建模的理论和应用。
光学活性分子材料,例如有机共轭聚合物和生物系统,其特征在于电子自由度和振动自由度之间的强耦合。通常,模拟必须超出Born-Oppenheimer逼近,才能说明激发态之间的非绝热耦合。实际上,非绝热动力学通常与激子动力学和光物理相关,涉及电荷和能量转移,以及激子解离和电荷复合。了解此类材料中的光致动力学对于提供激子形成,演化和衰减的准确描述至关重要。在过去的几十年中,这个跨学科领域已经显着成熟。制定新的理论框架,开发更高效,更准确的计算算法,随着高性能计算机硬件的发展,这些模拟已扩展到具有数百个原子的超大型分子系统,包括对有机半导体和生物分子的大量研究。在这篇综述中,我们将描述最近的理论进展,包括在表面跳跃方法中处理电子去相干,溶剂效应的作用,不可避免的琐碎交叉,基于跃迁密度的数据分析以及这些数值方法的有效计算实现。我们还强调了基于高斯近似的新开发的半经典方法,该方法保留了相位和宽度信息,以解决显着的相干和干扰效应,同时保持了表面跳变方法的高效率。


























 京公网安备 11010802027423号
京公网安备 11010802027423号