当前位置:
X-MOL 学术
›
Mol. Syst. Des. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Development of transferable coarse-grained models of amino acids
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2020-01-20 , DOI: 10.1039/c9me00173e Olivia Conway 1, 2, 3, 4 , Yaxin An 1, 2, 3, 4 , Karteek K. Bejagam 1, 2, 3, 4 , Sanket A. Deshmukh 1, 2, 3, 4
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2020-01-20 , DOI: 10.1039/c9me00173e Olivia Conway 1, 2, 3, 4 , Yaxin An 1, 2, 3, 4 , Karteek K. Bejagam 1, 2, 3, 4 , Sanket A. Deshmukh 1, 2, 3, 4
Affiliation
|
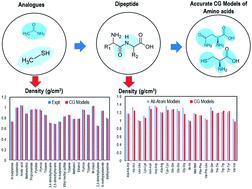
We have developed transferable coarse-grained (CG) models of the twenty standard amino acids, which can be used to perform molecular dynamics (MD) simulations of peptide amphiphiles (PAs) in the presence of explicit solvent. A 2 : 1 to 4 : 1 mapping scheme – in which a CG bead is comprised of two to four heavy atoms, respectively, and associated hydrogens – has been employed. Non-bonded parameters were optimized using the artificial neural network assisted particle swarm optimization (ANN-assisted PSO) method to reproduce experimental properties (density, surface tension, and heat of vaporization) of analogues of the side chains, termini, and backbone functional groups of the amino acids. The density (error <3.04%) and surface tension (error <7.38%) predicted by CG models were in good agreement with those of experimental properties. The peptide backbone is modeled with two charge neutral beads while amino acid side chains are modeled with one to three beads. Each terminus (N-terminus and C-terminus) is modeled as one charge neutral bead. Bonded parameters for the CG models were obtained from bond, angle, and dihedral distributions from AA MD simulations of dipeptides and/or tripeptides, which showed a reasonable agreement. Moreover, densities of these dipeptides and tripeptides calculated from AA MD simulations and CG models were in excellent agreement.
中文翻译:

氨基酸可转移粗粒度模型的开发
我们已经开发了二十种标准氨基酸的可转移粗粒度(CG)模型,可用于在显式溶剂存在下对肽两亲物(PA)进行分子动力学(MD)模拟。已经采用了2:1至4:1的映射方案(其中CG珠分别由2至4个重原子以及相关的氢组成)。使用人工神经网络辅助粒子群优化(ANN辅助PSO)方法优化非键合参数,以重现侧链,末端和骨架官能团类似物的实验性质(密度,表面张力和汽化热)氨基酸。CG模型预测的密度(误差<3.04%)和表面张力(误差<7.38%)与实验性质吻合良好。肽主链用两个带电荷的中性珠子模拟,而氨基酸侧链用一到三个珠子模拟。每个末端(N末端和C末端)被建模为一个电荷中性珠。CG模型的键合参数是从二肽和/或三肽的AA MD模拟的键合,角度和二面体分布获得的,这显示出合理的一致性。而且,由AA MD模拟和CG模型计算得到的这些二肽和三肽的密度非常一致。由二肽和/或三肽的AA MD模拟得出的二面角分布,显示出合理的一致性。而且,由AA MD模拟和CG模型计算得到的这些二肽和三肽的密度非常一致。由二肽和/或三肽的AA MD模拟得出的二面角分布,显示出合理的一致性。而且,由AA MD模拟和CG模型计算得到的这些二肽和三肽的密度非常一致。
更新日期:2020-01-20
中文翻译:
氨基酸可转移粗粒度模型的开发
我们已经开发了二十种标准氨基酸的可转移粗粒度(CG)模型,可用于在显式溶剂存在下对肽两亲物(PA)进行分子动力学(MD)模拟。已经采用了2:1至4:1的映射方案(其中CG珠分别由2至4个重原子以及相关的氢组成)。使用人工神经网络辅助粒子群优化(ANN辅助PSO)方法优化非键合参数,以重现侧链,末端和骨架官能团类似物的实验性质(密度,表面张力和汽化热)氨基酸。CG模型预测的密度(误差<3.04%)和表面张力(误差<7.38%)与实验性质吻合良好。肽主链用两个带电荷的中性珠子模拟,而氨基酸侧链用一到三个珠子模拟。每个末端(N末端和C末端)被建模为一个电荷中性珠。CG模型的键合参数是从二肽和/或三肽的AA MD模拟的键合,角度和二面体分布获得的,这显示出合理的一致性。而且,由AA MD模拟和CG模型计算得到的这些二肽和三肽的密度非常一致。由二肽和/或三肽的AA MD模拟得出的二面角分布,显示出合理的一致性。而且,由AA MD模拟和CG模型计算得到的这些二肽和三肽的密度非常一致。由二肽和/或三肽的AA MD模拟得出的二面角分布,显示出合理的一致性。而且,由AA MD模拟和CG模型计算得到的这些二肽和三肽的密度非常一致。










































 京公网安备 11010802027423号
京公网安备 11010802027423号