当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Integrating All-Atom and Coarse-Grained Simulations-Toward Understanding of IDPs at Surfaces.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-02-24 , DOI: 10.1021/acs.jctc.9b01041 Kristin Hyltegren 1 , Marco Polimeni 1 , Marie Skepö 1, 2 , Mikael Lund 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-02-24 , DOI: 10.1021/acs.jctc.9b01041 Kristin Hyltegren 1 , Marco Polimeni 1 , Marie Skepö 1, 2 , Mikael Lund 1, 2
Affiliation
|
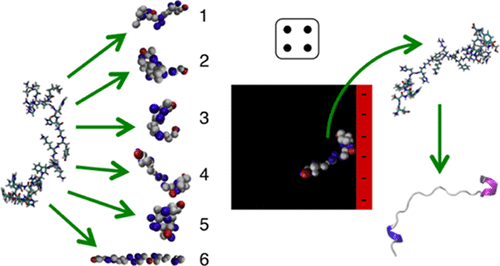
We present a scheme for transferring conformational degrees of freedom from all-atom (AA) simulations of an intrinsically disordered protein (IDP) to coarse-grained (CG) Monte Carlo (MC) simulations using conformational swap moves. AA simulations of a single histatin 5 peptide in water were used to obtain a structural ensemble, which is reweighted in a CGMC simulation in the presence of a negatively charged surface. For efficient sampling, the AA trajectory was condensed using two approaches: RMSD clustering (based on the root-mean-square difference in atom positions) and a "naı̈ve" truncation, where only every 100th frame of the trajectory was included in the library. The results show that even libraries with few structures well reproduce the radius of gyration and interaction free energy as functions of the distance from the surface. We further observe that the surface slightly promotes the secondary structure of histatin 5 and more so if using explicit surface charges rather than smeared charges.
中文翻译:

集成全原子和粗粒度模拟,以了解表面上的IDP。
我们提出了一种方案,用于将构象自由度从本质上无序的蛋白质(IDP)的所有原子(AA)模拟转移到使用构象交换移动的粗粒(CG)蒙特卡洛(MC)模拟。水中单个组蛋白5肽的AA模拟用于获得结构整体,在带负电荷的表面存在下通过CGMC模拟对其进行加权。为了进行有效采样,使用两种方法压缩了AA轨迹:RMSD聚类(基于原子位置的均方根差)和“幼稚”的截断,其中该轨迹的每100帧仅包含在库中。结果表明,即使是结构很少的库,也可以很好地重现回转半径和相互作用自由能,该自由度是与表面距离的函数。
更新日期:2020-02-24
中文翻译:
集成全原子和粗粒度模拟,以了解表面上的IDP。
我们提出了一种方案,用于将构象自由度从本质上无序的蛋白质(IDP)的所有原子(AA)模拟转移到使用构象交换移动的粗粒(CG)蒙特卡洛(MC)模拟。水中单个组蛋白5肽的AA模拟用于获得结构整体,在带负电荷的表面存在下通过CGMC模拟对其进行加权。为了进行有效采样,使用两种方法压缩了AA轨迹:RMSD聚类(基于原子位置的均方根差)和“幼稚”的截断,其中该轨迹的每100帧仅包含在库中。结果表明,即使是结构很少的库,也可以很好地重现回转半径和相互作用自由能,该自由度是与表面距离的函数。










































 京公网安备 11010802027423号
京公网安备 11010802027423号