当前位置:
X-MOL 学术
›
ChemMedChem
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Tranylcypromine-Based LSD1 Inhibitors: Structure-Activity Relationships, Antiproliferative Effects in Leukemia, and Gene Target Modulation.
ChemMedChem ( IF 3.6 ) Pub Date : 2020-01-31 , DOI: 10.1002/cmdc.201900730 Rossella Fioravanti 1 , Annalisa Romanelli 1 , Nicola Mautone 1 , Elisabetta Di Bello 1 , Annarita Rovere 1 , Davide Corinti 1 , Clemens Zwergel 1, 2 , Sergio Valente 1 , Dante Rotili 1 , Oronza A Botrugno 3, 4 , Paola Dessanti 3, 5 , Stefania Vultaggio 3 , Paola Vianello 3 , Anna Cappa 3, 6 , Claudia Binda 7 , Andrea Mattevi 7 , Saverio Minucci 3, 8 , Ciro Mercurio 3, 6 , Mario Varasi 3, 6 , Antonello Mai 1
ChemMedChem ( IF 3.6 ) Pub Date : 2020-01-31 , DOI: 10.1002/cmdc.201900730 Rossella Fioravanti 1 , Annalisa Romanelli 1 , Nicola Mautone 1 , Elisabetta Di Bello 1 , Annarita Rovere 1 , Davide Corinti 1 , Clemens Zwergel 1, 2 , Sergio Valente 1 , Dante Rotili 1 , Oronza A Botrugno 3, 4 , Paola Dessanti 3, 5 , Stefania Vultaggio 3 , Paola Vianello 3 , Anna Cappa 3, 6 , Claudia Binda 7 , Andrea Mattevi 7 , Saverio Minucci 3, 8 , Ciro Mercurio 3, 6 , Mario Varasi 3, 6 , Antonello Mai 1
Affiliation
|
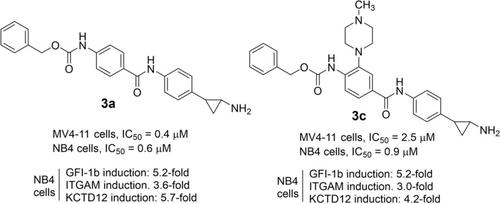
LSD1 is a lysine demethylase highly involved in initiation and development of cancer. To design highly effective covalent inhibitors, a strategy is to fill its large catalytic cleft by designing tranylcypromine (TCP) analogs decorated with long, hindered substituents. We prepared three series of TCP analogs, carrying aroyl- and arylacetylamino (1 a-h), Z-amino acylamino (2 a-o), or double-substituted benzamide (3 a-n) residues at the C4 or C3 position of the phenyl ring. Further fragments obtained by chemical manipulation applied on the TCP scaffold (compounds 4 a-i) were also prepared. When tested against LSD1, most of 1 and 3 exhibited IC50 values in the low nanomolar range, with 1 e and 3 a,d,f,g being also the most selective respect to monoamine oxidases. In MV4-11 AML and NB4 APL cells compounds 3 were the most potent, displaying up to sub-micromolar cell growth inhibition against both cell lines (3 a) or against NB4 cells (3 c). The most potent compounds in cellular assays were also able to induce the expression of LSD1 target genes, such as GFI-1b, ITGAM, and KCTD12, as functional read-out for LSD1 inhibition. Mouse and human intrinsic clearance data highlighted the high metabolic stability of compounds 3 a, 3 d and 3 g. Further studies will be performed on the new compounds 3 a and 3 c to assess their anticancer potential in different cancer contexts.
中文翻译:

基于Tranylcypromine的LSD1抑制剂:结构与活性的关系,白血病的抗增殖作用和基因靶标调控。
LSD1是赖氨酸脱甲基酶,高度参与癌症的发生和发展。为了设计高效的共价抑制剂,一种策略是通过设计装饰有长位受阻取代基的反式环丙胺(TCP)类似物来填补其大的催化裂隙。我们制备了三个系列的TCP类似物,在苯环的C4或C3位置带有芳酰基和芳基乙酰氨基(1 ah),Z-氨基酰基氨基(2 ao)或双取代的苯甲酰胺(3 an)残基。还制备了通过化学操作施加到TCP支架上的其他片段(化合物4ai)。当针对LSD1进行测试时,大多数1和3表现出的IC50值在低纳摩尔范围内,而1 e和3 a,d,f,g对单胺氧化酶的选择性也最高。在MV4-11 AML和NB4 APL细胞中,化合物3最有效,对两种细胞系(3a)或NB4细胞(3c)都表现出亚微摩尔细胞生长抑制作用。细胞测定中最有效的化合物还能够诱导LSD1靶基因(如GFI-1b,ITGAM和KCTD12)的表达,作为抑制LSD1的功能性读数。小鼠和人类的固有清除率数据突出了化合物3a,3d和3g的高代谢稳定性。将对新化合物3a和3c进行进一步研究,以评估其在不同癌症背景下的抗癌潜力。小鼠和人类的固有清除率数据突出了化合物3a,3d和3g的高代谢稳定性。将对新化合物3a和3c进行进一步研究,以评估其在不同癌症背景下的抗癌潜力。小鼠和人类的固有清除率数据突出了化合物3a,3d和3g的高代谢稳定性。将对新化合物3a和3c进行进一步研究,以评估其在不同癌症背景下的抗癌潜力。
更新日期:2020-02-14
中文翻译:
基于Tranylcypromine的LSD1抑制剂:结构与活性的关系,白血病的抗增殖作用和基因靶标调控。
LSD1是赖氨酸脱甲基酶,高度参与癌症的发生和发展。为了设计高效的共价抑制剂,一种策略是通过设计装饰有长位受阻取代基的反式环丙胺(TCP)类似物来填补其大的催化裂隙。我们制备了三个系列的TCP类似物,在苯环的C4或C3位置带有芳酰基和芳基乙酰氨基(1 ah),Z-氨基酰基氨基(2 ao)或双取代的苯甲酰胺(3 an)残基。还制备了通过化学操作施加到TCP支架上的其他片段(化合物4ai)。当针对LSD1进行测试时,大多数1和3表现出的IC50值在低纳摩尔范围内,而1 e和3 a,d,f,g对单胺氧化酶的选择性也最高。在MV4-11 AML和NB4 APL细胞中,化合物3最有效,对两种细胞系(3a)或NB4细胞(3c)都表现出亚微摩尔细胞生长抑制作用。细胞测定中最有效的化合物还能够诱导LSD1靶基因(如GFI-1b,ITGAM和KCTD12)的表达,作为抑制LSD1的功能性读数。小鼠和人类的固有清除率数据突出了化合物3a,3d和3g的高代谢稳定性。将对新化合物3a和3c进行进一步研究,以评估其在不同癌症背景下的抗癌潜力。小鼠和人类的固有清除率数据突出了化合物3a,3d和3g的高代谢稳定性。将对新化合物3a和3c进行进一步研究,以评估其在不同癌症背景下的抗癌潜力。小鼠和人类的固有清除率数据突出了化合物3a,3d和3g的高代谢稳定性。将对新化合物3a和3c进行进一步研究,以评估其在不同癌症背景下的抗癌潜力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号