当前位置:
X-MOL 学术
›
Eur. J. Hum. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Genotype phasing in pedigrees using whole-genome sequence data.
European Journal of Human Genetics ( IF 3.7 ) Pub Date : 2020-01-29 , DOI: 10.1038/s41431-020-0574-3 August N Blackburn 1, 2 , Lucy Blondell 1 , Mark Z Kos 1 , Nicholas B Blackburn 1 , Juan M Peralta 1 , Peter T Stevens 1 , Donna M Lehman 3 , John Blangero 1 , Harald H H Göring 1
European Journal of Human Genetics ( IF 3.7 ) Pub Date : 2020-01-29 , DOI: 10.1038/s41431-020-0574-3 August N Blackburn 1, 2 , Lucy Blondell 1 , Mark Z Kos 1 , Nicholas B Blackburn 1 , Juan M Peralta 1 , Peter T Stevens 1 , Donna M Lehman 3 , John Blangero 1 , Harald H H Göring 1
Affiliation
|
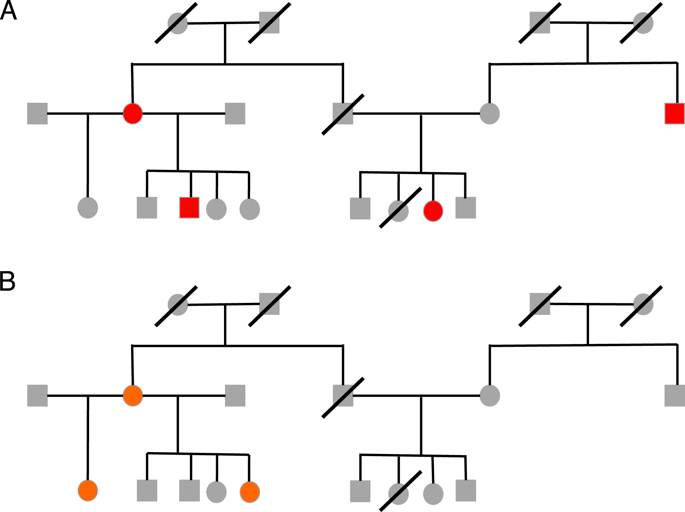
Phasing is the process of inferring haplotypes from genotype data. Efficient algorithms and associated software for accurate phasing in pedigrees are needed, especially for populations lacking reference panels of sequenced individuals. We present a novel method for phasing genotypes from whole-genome sequence data in pedigrees, called PULSAR (Phasing Using Lineage Specific Alleles/Rare variants). The method is based on the property that alleles specific to a single founding chromosome within a pedigree are highly informative for identifying haplotypes that are shared identical by descent. Simulation studies are used to assess the performance of PULSAR with various pedigree sizes and structures, and the effect of genotyping errors and the presence of nonsequenced individuals is investigated. In pedigrees with complete sequencing and realistic genotyping error rates, PULSAR correctly phases >99.9% of heterozygous genotypes, excluding sites at which all individuals are heterozygous, and does so with a switch error rate frequently below 10-4. PULSAR is highly accurate, capable of genotype error correction and imputation, and computationally competitive with alternative phasing software applicable to pedigrees. Our method has the significant advantage of not requiring reference panels that are essential for other population-based phasing algorithms. A software implementation of PULSAR is freely available.
中文翻译:

使用全基因组序列数据在谱系中进行基因型定相。
分阶段是根据基因型数据推断单倍型的过程。需要用于谱系中准确定相的高效算法和相关软件,尤其是对于缺少已测序个体参考面板的人群。我们提出了一种从谱系中全基因组序列数据中分型基因型的新方法,称为PULSAR(使用谱系特异性等位基因/稀有变体进行分期)。该方法基于这样的特性,即血统中特定于单个创始染色体的等位基因对于鉴定由血统共享相同的单倍型具有很高的信息价值。仿真研究被用于评估不同谱系大小和结构的PULSAR的性能,并研究了基因分型错误和未测序个体的存在的影响。在具有完整测序和实际基因分型错误率的谱系中,PULSAR正确地定相> 99.9%的杂合基因型,排除所有个体都是杂合的位点,并且这样做的转换错误率经常低于10-4。PULSAR高度准确,能够进行基因型错误校正和估算,并且与适用于谱系的替代定相软件在计算上具有竞争力。我们的方法的显着优势是不需要参考面板,而其他参考面板是其他基于人口的定相算法所必需的。PULSAR的软件实现是免费提供的。能够进行基因型错误校正和归因,并且在计算上与适用于谱系的替代定相软件具有竞争力。我们的方法的显着优势是不需要参考面板,而其他参考面板是其他基于人口的定相算法所必需的。PULSAR的软件实现是免费提供的。能够进行基因型错误校正和归因,并且在计算上与适用于谱系的替代定相软件具有竞争力。我们的方法的显着优势是不需要参考面板,而其他参考面板是其他基于人口的定相算法所必需的。PULSAR的软件实现是免费提供的。
更新日期:2020-01-29
中文翻译:
使用全基因组序列数据在谱系中进行基因型定相。
分阶段是根据基因型数据推断单倍型的过程。需要用于谱系中准确定相的高效算法和相关软件,尤其是对于缺少已测序个体参考面板的人群。我们提出了一种从谱系中全基因组序列数据中分型基因型的新方法,称为PULSAR(使用谱系特异性等位基因/稀有变体进行分期)。该方法基于这样的特性,即血统中特定于单个创始染色体的等位基因对于鉴定由血统共享相同的单倍型具有很高的信息价值。仿真研究被用于评估不同谱系大小和结构的PULSAR的性能,并研究了基因分型错误和未测序个体的存在的影响。在具有完整测序和实际基因分型错误率的谱系中,PULSAR正确地定相> 99.9%的杂合基因型,排除所有个体都是杂合的位点,并且这样做的转换错误率经常低于10-4。PULSAR高度准确,能够进行基因型错误校正和估算,并且与适用于谱系的替代定相软件在计算上具有竞争力。我们的方法的显着优势是不需要参考面板,而其他参考面板是其他基于人口的定相算法所必需的。PULSAR的软件实现是免费提供的。能够进行基因型错误校正和归因,并且在计算上与适用于谱系的替代定相软件具有竞争力。我们的方法的显着优势是不需要参考面板,而其他参考面板是其他基于人口的定相算法所必需的。PULSAR的软件实现是免费提供的。能够进行基因型错误校正和归因,并且在计算上与适用于谱系的替代定相软件具有竞争力。我们的方法的显着优势是不需要参考面板,而其他参考面板是其他基于人口的定相算法所必需的。PULSAR的软件实现是免费提供的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号