当前位置:
X-MOL 学术
›
Bioorgan. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structure-based design and optimization of pyrimidine- and 1,2,4-triazolo[4,3-a]pyrimidine-based matrix metalloproteinase-10/13 inhibitors via Dimroth rearrangement towards targeted polypharmacology.
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2020-01-25 , DOI: 10.1016/j.bioorg.2020.103616 El Sayed Helmy El Ashry 1 , Laila Fathy Awad 1 , Mohamed Teleb 2 , Nihal Ahmed Ibrahim 1 , Marwa M Abu-Serie 3 , Mohamed Nabil Abd Al Moaty 1
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2020-01-25 , DOI: 10.1016/j.bioorg.2020.103616 El Sayed Helmy El Ashry 1 , Laila Fathy Awad 1 , Mohamed Teleb 2 , Nihal Ahmed Ibrahim 1 , Marwa M Abu-Serie 3 , Mohamed Nabil Abd Al Moaty 1
Affiliation
|
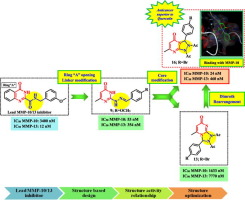
Recently, interest in matrix metalloproteinases (MMPs) -10 and -13 has been revitalized with the growing knowledge on their relevance within the MMPs network and significance of their inhibition for treatment of various diseases like arthritis, cancer, atherosclerosis and Alzheimer. Within this approach, dual MMP-10/13 inhibition was disclosed as new approach for targeted polypharmacology. While several efficient MMP-13 inhibitors are known, very few potent and selective MMP-10 inhibitors were reported. This study describes the design, synthesis and optimization of novel MMP-10/13 inhibitors with enhanced MMP-10 potency and selectivity towards polypharmacology. Starting with a lead fused pyrimidine-based MMP-13 inhibitor with weak MMP-10 inhibition, a structure-based design of pyrimidine and fused pyrimidine scaffolds was rationalized to enhance activity against MMP-10 in parallel with MMP-13. Firstly, a series of 6-methyl pyrimidin-4-one hydrazones 6-10 was synthesized via conventional and ultrasonic-assisted methods, then evaluated for MMP-10/13 inhibition. The most active derivative 9 exhibited acceptable dual potency with 7-fold selectivity for MMP-10 (IC50 = 53 nM) over MMP-13. Such hydrazones were then cyclized to the corresponding isomeric 1,2,4-triazolo[4,3-a]pyrimidines 12-19. Their MMP-10/13 inhibition assay revealed, in most cases, superior dual activities with general MMP-10 selectivity compared to the corresponding precursors 6-10. In addition, a clear structure activity relationship trend was deduced within the identified regioisomers, where the 5-oxo-1,2,4-triazolo[4,3-a]pyrimidine derivatives 15 and 16 were far more active against MMP-10/13 than their regioisomers 12 and 13. Remarkably, the p-bromophenyl derivative 16 exhibited the highest MMP-10 inhibition (IC50 = 24 nM), whereas the p-methoxy derivative 18 was the most potent MMP-13 inhibitor (IC50 = 294 nM). Moreover, 16 exhibited 19-fold selectivity for MMP-10 over MMP-13, 10-fold over MMP-9, and 29-fold over MMP-7. Docking studies were performed to provide reasonable explanation for structure-activity relationships and isoform selectivity. 16 and 18 were then evaluated for their anticancer activities against three human cancers to assess their therapeutic potential at cellular level via MTT assay. Both compounds exhibited superior anticancer activities compared to quercetin. Their in silico ligand efficiency metrics, physicochemical properties and ADME parameters were drug-like. Guided by such findings that point to 16 as the most promising compound in this study, further structure optimization was carried out via photoirradiation-mediated Dimroth rearrangement of the inactive triazolopyrimidine 13 to its potent regioisomer 16.
中文翻译:

基于结构的设计和嘧啶和1,2,4-三唑并[4,3-a]嘧啶类基质金属蛋白酶-10/13抑制剂通过Dimroth重排向目标多药理学优化。
最近,随着人们对基质金属蛋白酶(MMPs)-10和-13在基质金属蛋白酶(MMPs)网络中的相关性及其对多种疾病(如关节炎,癌症,动脉粥样硬化和阿尔茨海默氏病)的抑制作用的认识不断增强,人们对基质金属蛋白酶(MMPs)-10和-13的兴趣日益浓厚。在这种方法中,双重MMP-10 / 13抑制被公开为靶向多药理学的新方法。尽管已知几种有效的MMP-13抑制剂,但报道的有效和选择性MMP-10抑制剂很少。这项研究描述了新型MMP-10 / 13抑制剂的设计,合成和优化,这些抑制剂具有增强的MMP-10效力和对多药理学的选择性。从对MMP-10抑制作用较弱的基于铅融合的嘧啶的MMP-13抑制剂开始,合理设计了基于嘧啶和嘧啶融合支架的结构,以增强与MMP-13平行的抗MMP-10活性。首先,通过常规和超声辅助方法合成了一系列6-甲基嘧啶-4-酮6-10,然后评估了其对MMP-10 / 13的抑制作用。最具活性的衍生物9表现出可接受的双重效力,对MMP-10的选择性是MMP-13的7倍(IC50 = 53 nM)。然后将这种环环化为相应的异构体1,2,4-三唑并[4,3-a]嘧啶12-19。他们的MMP-10 / 13抑制试验显示,在大多数情况下,与相应的前体6-10相比,具有一般的MMP-10选择性更高的双重活性。此外,在确定的区域异构体中推导出了明确的结构活性关系趋势,其中5-氧代1,2,4-三唑[4,3-a]嘧啶衍生物15和16对MMP-10 / 13的活性远高于其区域异构体12和13。值得注意的是,对溴苯基衍生物16表现出最高的MMP-10抑制作用(IC50 = 24 nM),而对甲氧基衍生物18是最有效的MMP-13抑制剂(IC50 = 294 nM)。此外,有16种化合物对MMP-10的选择性是MMP-13的19倍,MMP-9的选择性是10倍,MMP-7的选择性是29倍。进行了对接研究,以提供结构活性关系和同工型选择性的合理解释。然后评估16和18对三种人类癌症的抗癌活性,以通过MTT分析在细胞水平评估其治疗潜力。与槲皮素相比,这两种化合物均显示出优异的抗癌活性。他们的计算机配体效率指标,理化性质和ADME参数呈药物样。在这些发现中指出16是本研究中最有希望的化合物,通过无活性三唑并嘧啶13的光辐照介导的Dimroth重排为其有效的区域异构体16进行了进一步的结构优化。
更新日期:2020-01-26
中文翻译:
基于结构的设计和嘧啶和1,2,4-三唑并[4,3-a]嘧啶类基质金属蛋白酶-10/13抑制剂通过Dimroth重排向目标多药理学优化。
最近,随着人们对基质金属蛋白酶(MMPs)-10和-13在基质金属蛋白酶(MMPs)网络中的相关性及其对多种疾病(如关节炎,癌症,动脉粥样硬化和阿尔茨海默氏病)的抑制作用的认识不断增强,人们对基质金属蛋白酶(MMPs)-10和-13的兴趣日益浓厚。在这种方法中,双重MMP-10 / 13抑制被公开为靶向多药理学的新方法。尽管已知几种有效的MMP-13抑制剂,但报道的有效和选择性MMP-10抑制剂很少。这项研究描述了新型MMP-10 / 13抑制剂的设计,合成和优化,这些抑制剂具有增强的MMP-10效力和对多药理学的选择性。从对MMP-10抑制作用较弱的基于铅融合的嘧啶的MMP-13抑制剂开始,合理设计了基于嘧啶和嘧啶融合支架的结构,以增强与MMP-13平行的抗MMP-10活性。首先,通过常规和超声辅助方法合成了一系列6-甲基嘧啶-4-酮6-10,然后评估了其对MMP-10 / 13的抑制作用。最具活性的衍生物9表现出可接受的双重效力,对MMP-10的选择性是MMP-13的7倍(IC50 = 53 nM)。然后将这种环环化为相应的异构体1,2,4-三唑并[4,3-a]嘧啶12-19。他们的MMP-10 / 13抑制试验显示,在大多数情况下,与相应的前体6-10相比,具有一般的MMP-10选择性更高的双重活性。此外,在确定的区域异构体中推导出了明确的结构活性关系趋势,其中5-氧代1,2,4-三唑[4,3-a]嘧啶衍生物15和16对MMP-10 / 13的活性远高于其区域异构体12和13。值得注意的是,对溴苯基衍生物16表现出最高的MMP-10抑制作用(IC50 = 24 nM),而对甲氧基衍生物18是最有效的MMP-13抑制剂(IC50 = 294 nM)。此外,有16种化合物对MMP-10的选择性是MMP-13的19倍,MMP-9的选择性是10倍,MMP-7的选择性是29倍。进行了对接研究,以提供结构活性关系和同工型选择性的合理解释。然后评估16和18对三种人类癌症的抗癌活性,以通过MTT分析在细胞水平评估其治疗潜力。与槲皮素相比,这两种化合物均显示出优异的抗癌活性。他们的计算机配体效率指标,理化性质和ADME参数呈药物样。在这些发现中指出16是本研究中最有希望的化合物,通过无活性三唑并嘧啶13的光辐照介导的Dimroth重排为其有效的区域异构体16进行了进一步的结构优化。










































 京公网安备 11010802027423号
京公网安备 11010802027423号