当前位置:
X-MOL 学术
›
J. Chem. Thermodyn.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Studies on the effect of temperature on dielectric relaxation, activation energy (ΔG*), enthalpy (ΔH*), entropy (ΔS*) and molecular interactions of some anilines, phenol and their binary mixtures using X-band microwave bench
The Journal of Chemical Thermodynamics ( IF 2.2 ) Pub Date : 2020-05-01 , DOI: 10.1016/j.jct.2020.106068 C.V. Maridevarmath , G.H. Malimath
The Journal of Chemical Thermodynamics ( IF 2.2 ) Pub Date : 2020-05-01 , DOI: 10.1016/j.jct.2020.106068 C.V. Maridevarmath , G.H. Malimath
|
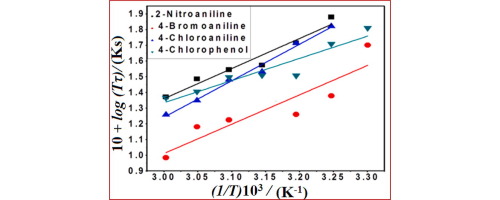
Abstract In the present study, the variation of dielectric relaxation time (τ) with temperature is carried out on four polar molecules viz., 2-Nitroaniline, 4-Bromoaniline, 4-Chloroaniline and 4-Chlorophenol and also on the binary mixtures (1:1) viz., (2-Nitroaniline + 4-Bromoaniline) and (4-Chloroaniline + 4-Chlorophenol) in benzene and results are analysed using Whiffen and Thompson model in order to understand the nature of the relaxation process involved. Microwave bench operating at a frequency of 9.59 GHz is used for this purpose. It is observed that, relaxation time (τ) decreases with increase in temperature for all the systems studied and the results are in agreement with Debye’s theory. The linearity in the log (τT) versus 1/T plots for all the cases indicates that, relaxation process can be treated as rate process according to Eyring’s theory. Further, thermodynamic parameters like change in activation energy (ΔG*), enthalpy of activation (ΔH*) and entropy of activation (ΔS*) have been determined for all the cases. It is observed that, values of ΔH* are higher than the values of ΔG*, thus leading to the positive values of ΔS*. The positive values of ΔS* indicates that, the environment of the system is non-cooperative for the activated process and the activated state is less stable and disordered than the normal state for all the cases studied. The molecular radii estimated from SED theory, Edward’s atomic increment method and from DFT computations using Gaussian 09 W are found to be in good agreement with each other and suggest that, the molecular dynamics follows SED theory.
中文翻译:

使用 X 波段微波台架研究温度对某些苯胺、苯酚及其二元混合物的介电弛豫、活化能 (ΔG*)、焓 (ΔH*)、熵 (ΔS*) 和分子相互作用的影响
摘要 在本研究中,介电弛豫时间 (τ) 随温度的变化在四种极性分子上进行,即 2-硝基苯胺、4-溴苯胺、4-氯苯胺和 4-氯苯酚以及二元混合物 (1 :1) 即苯中的(2-硝基苯胺 + 4-溴苯胺)和(4-氯苯胺 + 4-氯苯酚),并使用 Whiffen 和 Thompson 模型分析结果,以了解所涉及的松弛过程的性质。为此,使用频率为 9.59 GHz 的微波工作台。观察到,对于所有研究的系统,弛豫时间(τ)随着温度的升高而减少,结果与德拜的理论一致。所有情况的对数 (τT) 与 1/T 图中的线性关系表明,根据Eyring的理论,松弛过程可以看作是速率过程。此外,已经确定了所有情况下的热力学参数,如活化能 (ΔG*) 的变化、活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。已经确定了所有情况下的热力学参数,如活化能变化 (ΔG*)、活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。已经确定了所有情况下的热力学参数,如活化能变化 (ΔG*)、活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。已确定所有情况的活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。已确定所有情况的活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。
更新日期:2020-05-01
中文翻译:
使用 X 波段微波台架研究温度对某些苯胺、苯酚及其二元混合物的介电弛豫、活化能 (ΔG*)、焓 (ΔH*)、熵 (ΔS*) 和分子相互作用的影响
摘要 在本研究中,介电弛豫时间 (τ) 随温度的变化在四种极性分子上进行,即 2-硝基苯胺、4-溴苯胺、4-氯苯胺和 4-氯苯酚以及二元混合物 (1 :1) 即苯中的(2-硝基苯胺 + 4-溴苯胺)和(4-氯苯胺 + 4-氯苯酚),并使用 Whiffen 和 Thompson 模型分析结果,以了解所涉及的松弛过程的性质。为此,使用频率为 9.59 GHz 的微波工作台。观察到,对于所有研究的系统,弛豫时间(τ)随着温度的升高而减少,结果与德拜的理论一致。所有情况的对数 (τT) 与 1/T 图中的线性关系表明,根据Eyring的理论,松弛过程可以看作是速率过程。此外,已经确定了所有情况下的热力学参数,如活化能 (ΔG*) 的变化、活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。已经确定了所有情况下的热力学参数,如活化能变化 (ΔG*)、活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。已经确定了所有情况下的热力学参数,如活化能变化 (ΔG*)、活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。已确定所有情况的活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。已确定所有情况的活化焓 (ΔH*) 和活化熵 (ΔS*)。观察到,ΔH*的值高于ΔG*的值,从而导致ΔS*的正值。ΔS* 的正值表明,系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。系统的环境对于激活过程是非合作的,并且在所有研究的情况下,激活状态比正常状态更不稳定和无序。发现根据 SED 理论、Edward 的原子增量方法和使用 Gaussian 09 W 的 DFT 计算估计的分子半径彼此非常吻合,表明分子动力学遵循 SED 理论。










































 京公网安备 11010802027423号
京公网安备 11010802027423号