Acta Pharmacologica Sinica ( IF 6.9 ) Pub Date : 2020-01-22 , DOI: 10.1038/s41401-019-0353-2 Wei-Min Kong 1 , Bin-Bin Sun 1 , Zhong-Jian Wang 1 , Xiao-Ke Zheng 1 , Kai-Jing Zhao 1 , Yang Chen 1 , Jia-Xin Zhang 1 , Pei-Hua Liu 1 , Liang Zhu 1 , Ru-Jun Xu 1 , Ping Li 1 , Li Liu 1 , Xiao-Dong Liu 1
|
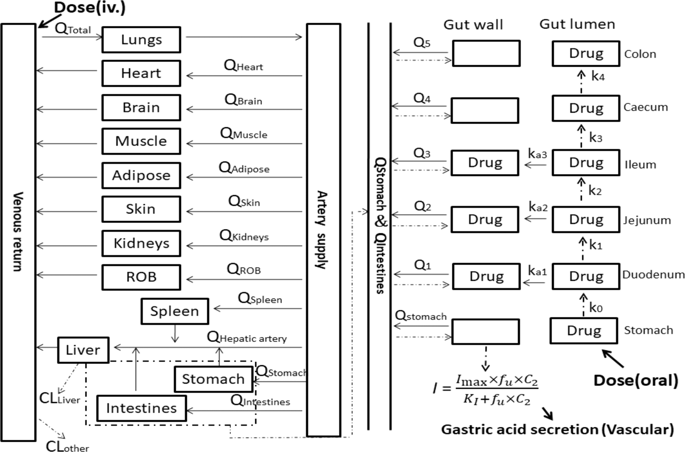
Vonoprazan is characterized as having a long-lasting antisecretory effect on gastric acid. In this study we developed a physiologically based pharmacokinetic (PBPK)-pharmacodynamic (PD) model linking to stomach to simultaneously predict vonoprazan pharmacokinetics and its antisecretory effects following administration to rats, dogs, and humans based on in vitro parameters. The vonoprazan disposition in the stomach was illustrated using a limited-membrane model. In vitro metabolic and transport parameters were derived from hepatic microsomes and Caco-2 cells, respectively. We found the most predicted plasma concentrations and pharmacokinetic parameters of vonoprazan in rats, dogs and humans were within twofold errors of the observed data. Free vonoprazan concentrations (fu × C2) in the stomach were simulated and linked to the antisecretory effects of the drug (I) (increases in pH or acid output) using the fomula dI/dt = k × fu × C2 × (Imax − I) − kd × I. The vonoprazan dissociation rate constant kd (0.00246 min−1) and inhibition index KI (35 nM) for H+/K+-ATPase were obtained from literatures. The vonoprazan-H+/K+-ATPase binding rate constant k was 0.07028 min−1· μM−1 using ratio of kd to KI. The predicted antisecretory effects were consistent with the observations following intravenous administration to rats (0.7 and 1.0 mg/kg), oral administration to dogs (0.3 and 1.0 mg/kg) and oral single dose or multidose to humans (20, 30, and 40 mg). Simulations showed that vonoprazan concentrations in stomach were 1000-fold higher than those in the plasma at 24 h following administration to human. Vonoprazan pharmacokinetics and its antisecretory effects may be predicted from in vitro data using the PBPK-PD model of the stomach. These findings may highlight 24-h antisecretory effects of vonoprazan in humans following single-dose or the sustained inhibition throughout each 24-h dosing interval during multidose administration.
中文翻译:
基于生理学的药代动力学-药效学模型,用于预测大鼠、狗和人类静脉/口服给药后沃诺拉赞的药代动力学及其对胃酸分泌的抑制作用
沃诺拉赞的特点是对胃酸具有持久的抗分泌作用。在这项研究中,我们开发了一种与胃相关的基于生理的药代动力学 (PBPK)-药效学 (PD) 模型,以根据体外参数同时预测沃诺拉赞的药代动力学及其对大鼠、狗和人类给药后的抗分泌作用。使用有限膜模型说明了胃中的沃诺拉赞分布。体外代谢和转运参数分别来自肝微粒体和 Caco-2 细胞。我们发现,在大鼠、狗和人类中,vonoprazan 的最预测血浆浓度和药代动力学参数在观察数据的两倍误差范围内。游离沃诺拉赞浓度 ( f u × C2 ) 使用公式dI / dt = k × f u × C 2 × ( I max - I ) - k模拟胃中的药物 ( I ) (增加 pH 或酸输出)的抗分泌作用d × 我。H + /K + -ATP酶的vonoprazan解离速率常数k d (0.00246 min -1 )和抑制指数K I (35 nM)从文献中获得。vonoprazan-H + /K+ -ATPase 结合速率常数k为 0.07028 min -1 · μM -1使用k d与K I的比率. 预测的抗分泌作用与大鼠静脉给药(0.7 和 1.0 mg/kg)、狗口服给药(0.3 和 1.0 mg/kg)和人口服单剂量或多剂量(20、30 和 40毫克)。模拟表明,在人体给药后 24 小时,胃中的沃诺拉赞浓度比血浆中的浓度高 1000 倍。使用胃的 PBPK-PD 模型,可以从体外数据预测 Vonoprazan 的药代动力学及其抗分泌作用。这些发现可能突出了沃诺拉赞在单次给药后对人体的 24 小时抗分泌作用,或在多剂量给药期间在每个 24 小时给药间隔内的持续抑制作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号