Nature ( IF 50.5 ) Pub Date : 2020-01-22 , DOI: 10.1038/s41586-020-1947-z Guoxia Liu 1 , Arianne Papa 2 , Alexander N Katchman 1 , Sergey I Zakharov 1 , Daniel Roybal 3 , Jessica A Hennessey 1 , Jared Kushner 1 , Lin Yang 1 , Bi-Xing Chen 1 , Alexander Kushnir 1 , Katerina Dangas 1 , Steven P Gygi 4 , Geoffrey S Pitt 5 , Henry M Colecraft 2, 3 , Manu Ben-Johny 2 , Marian Kalocsay 6 , Steven O Marx 1, 3
|
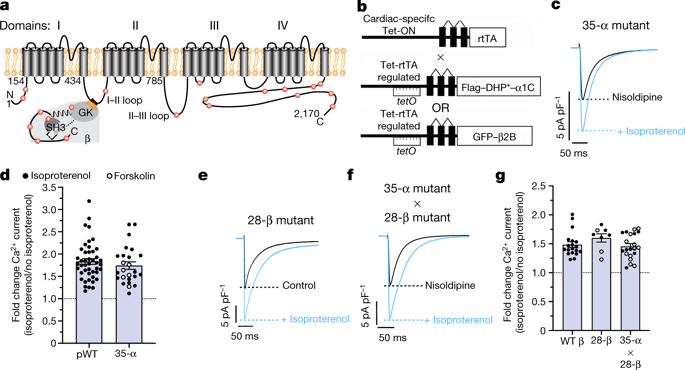
Increased cardiac contractility during the fight-or-flight response is caused by β-adrenergic augmentation of CaV1.2 voltage-gated calcium channels1,2,3,4. However, this augmentation persists in transgenic murine hearts expressing mutant CaV1.2 α1C and β subunits that can no longer be phosphorylated by protein kinase A—an essential downstream mediator of β-adrenergic signalling—suggesting that non-channel factors are also required. Here we identify the mechanism by which β-adrenergic agonists stimulate voltage-gated calcium channels. We express α1C or β2B subunits conjugated to ascorbate peroxidase5 in mouse hearts, and use multiplexed quantitative proteomics6,7 to track hundreds of proteins in the proximity of CaV1.2. We observe that the calcium-channel inhibitor Rad8,9, a monomeric G protein, is enriched in the CaV1.2 microenvironment but is depleted during β-adrenergic stimulation. Phosphorylation by protein kinase A of specific serine residues on Rad decreases its affinity for β subunits and relieves constitutive inhibition of CaV1.2, observed as an increase in channel open probability. Expression of Rad or its homologue Rem in HEK293T cells also imparts stimulation of CaV1.3 and CaV2.2 by protein kinase A, revealing an evolutionarily conserved mechanism that confers adrenergic modulation upon voltage-gated calcium channels.
中文翻译:
邻近蛋白质组学揭示肾上腺素CaV1.2刺激机制
战斗或逃跑反应期间心肌收缩力增加是由 Ca V 1.2 电压门控钙通道1,2,3,4的 β-肾上腺素能增强引起的。然而,这种增强作用在表达突变 Ca V 1.2 α 1C和 β 亚基的转基因小鼠心脏中持续存在,这些亚基不再被蛋白激酶 A(β-肾上腺素信号传导的重要下游介质)磷酸化,这表明还需要非通道因子。在这里,我们确定了 β-肾上腺素能激动剂刺激电压门控钙通道的机制。我们在小鼠心脏中表达与抗坏血酸过氧化物酶5缀合的 α 1C或 β 2B亚基,并使用多重定量蛋白质组学6,7追踪 Ca V 1.2 附近的数百种蛋白质。我们观察到钙通道抑制剂 Rad 8,9是一种单体 G 蛋白,在 Ca V 1.2 微环境中富集,但在 β-肾上腺素能刺激过程中被耗尽。蛋白激酶 A 对 Rad 上特定丝氨酸残基的磷酸化降低了其对 β 亚基的亲和力,并减轻了 Ca V 1.2 的组成型抑制,观察到通道开放概率增加。 HEK293T 细胞中 Rad 或其同源物 Rem 的表达也会通过蛋白激酶 A 刺激 Ca V 1.3 和 Ca V 2.2,揭示了一种进化上保守的机制,可对电压门控钙通道进行肾上腺素能调节。










































 京公网安备 11010802027423号
京公网安备 11010802027423号