当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Semiempirical methods do Fukui functions: Unlocking a modeling framework for biosystems
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-01-20 , DOI: 10.1002/jcc.26148 Igor Barden Grillo 1 , Gabriel A Urquiza-Carvalho 2 , Elton José Ferreira Chaves 3 , Gerd Bruno Rocha 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-01-20 , DOI: 10.1002/jcc.26148 Igor Barden Grillo 1 , Gabriel A Urquiza-Carvalho 2 , Elton José Ferreira Chaves 3 , Gerd Bruno Rocha 1
Affiliation
|
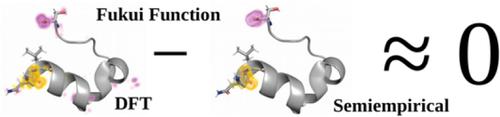
Obtaining reactivity information from the molecular electronic structure of a chemical system is a computationally intensive process. As a way of probing reactivity information around that, there exist electron density response variables, such as the Fukui functions (FFs), which are well‐established descriptors that summarize the local susceptibility to react. These properties only require few single‐point quantum chemical calculations, but even then, the intrinsic high cost and unfavorable computational complexity with respect to the number of atoms in the system makes this approach available only to small fragments and systems. In this study, we explore the computation of FFs, showing that semiempirical quantum chemical methods can be used to obtain the reactivity information equivalent to that of a Density Functional Theory (DFT) functional, for the eight entire polypeptide chains. The combination of semiempirical methods with the frozen orbital approximation allows for the obtention of these reactivity descriptors for biological systems with reasonable accuracy and speed, unlocking the utilization of these methods for such systems. These results for the frozen orbital approximation can be additionally improved when other molecular orbitals from the frontier band are employed in the computation. We also show the potential of this computational protocol in the ligand–protein complexes of HIV‐1 protease, predicting which of those ligands are active inhibitors.
中文翻译:

半经验方法做福井函数:解锁生物系统的建模框架
从化学系统的分子电子结构中获取反应性信息是一个计算密集型过程。作为探索反应性信息的一种方式,存在电子密度响应变量,例如 Fukui 函数 (FFs),它们是总结局部反应敏感性的成熟描述符。这些特性只需要很少的单点量子化学计算,但即便如此,与系统中原子数量相关的固有高成本和不利的计算复杂性使得这种方法仅适用于小片段和系统。在这项研究中,我们探索了 FF 的计算,表明半经验量子化学方法可用于获得等效于密度泛函理论 (DFT) 泛函的反应性信息,对于八个完整的多肽链。半经验方法与冻结轨道近似的结合允许以合理的精度和速度获得生物系统的这些反应性描述符,从而解锁这些方法对此类系统的利用。当在计算中使用来自前沿带的其他分子轨道时,可以进一步改进冻结轨道近似的这些结果。我们还展示了这种计算方案在 HIV-1 蛋白酶的配体-蛋白质复合物中的潜力,预测哪些配体是活性抑制剂。半经验方法与冻结轨道近似的结合允许以合理的精度和速度获得生物系统的这些反应性描述符,从而解锁这些方法对此类系统的利用。当在计算中使用来自前沿带的其他分子轨道时,可以进一步改进冻结轨道近似的这些结果。我们还展示了这种计算方案在 HIV-1 蛋白酶的配体-蛋白质复合物中的潜力,预测哪些配体是活性抑制剂。半经验方法与冻结轨道近似的结合允许以合理的精度和速度获得生物系统的这些反应性描述符,从而解锁这些方法对此类系统的利用。当在计算中使用来自前沿带的其他分子轨道时,可以进一步改进冻结轨道近似的这些结果。我们还展示了这种计算方案在 HIV-1 蛋白酶的配体-蛋白质复合物中的潜力,预测哪些配体是活性抑制剂。
更新日期:2020-01-20
中文翻译:
半经验方法做福井函数:解锁生物系统的建模框架
从化学系统的分子电子结构中获取反应性信息是一个计算密集型过程。作为探索反应性信息的一种方式,存在电子密度响应变量,例如 Fukui 函数 (FFs),它们是总结局部反应敏感性的成熟描述符。这些特性只需要很少的单点量子化学计算,但即便如此,与系统中原子数量相关的固有高成本和不利的计算复杂性使得这种方法仅适用于小片段和系统。在这项研究中,我们探索了 FF 的计算,表明半经验量子化学方法可用于获得等效于密度泛函理论 (DFT) 泛函的反应性信息,对于八个完整的多肽链。半经验方法与冻结轨道近似的结合允许以合理的精度和速度获得生物系统的这些反应性描述符,从而解锁这些方法对此类系统的利用。当在计算中使用来自前沿带的其他分子轨道时,可以进一步改进冻结轨道近似的这些结果。我们还展示了这种计算方案在 HIV-1 蛋白酶的配体-蛋白质复合物中的潜力,预测哪些配体是活性抑制剂。半经验方法与冻结轨道近似的结合允许以合理的精度和速度获得生物系统的这些反应性描述符,从而解锁这些方法对此类系统的利用。当在计算中使用来自前沿带的其他分子轨道时,可以进一步改进冻结轨道近似的这些结果。我们还展示了这种计算方案在 HIV-1 蛋白酶的配体-蛋白质复合物中的潜力,预测哪些配体是活性抑制剂。半经验方法与冻结轨道近似的结合允许以合理的精度和速度获得生物系统的这些反应性描述符,从而解锁这些方法对此类系统的利用。当在计算中使用来自前沿带的其他分子轨道时,可以进一步改进冻结轨道近似的这些结果。我们还展示了这种计算方案在 HIV-1 蛋白酶的配体-蛋白质复合物中的潜力,预测哪些配体是活性抑制剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号