当前位置:
X-MOL 学术
›
Cell Death Differ.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Prolyl hydroxylase 3 controls the intestine goblet cell generation through stabilizing ATOH1.
Cell Death and Differentiation ( IF 13.7 ) Pub Date : 2020-01-20 , DOI: 10.1038/s41418-020-0490-7 Yi-Ming Xu 1 , Qiang Gao 1 , Jin-Zhao Zhang 1 , Yun-Tao Lu 1 , Dong-Ming Xing 2, 3 , Yan-Qing Qin 1 , Jing Fang 2, 3
Cell Death and Differentiation ( IF 13.7 ) Pub Date : 2020-01-20 , DOI: 10.1038/s41418-020-0490-7 Yi-Ming Xu 1 , Qiang Gao 1 , Jin-Zhao Zhang 1 , Yun-Tao Lu 1 , Dong-Ming Xing 2, 3 , Yan-Qing Qin 1 , Jing Fang 2, 3
Affiliation
|
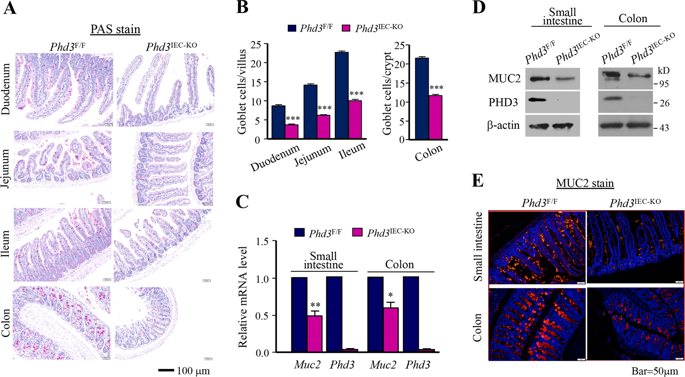
Intestinal epithelia self-renew constantly and generate differentiated cells such as secretary goblet cells. The intestine goblet cells secrete gel-forming mucins that form mucus to create a barrier of defense. We reported previously that loss of prolyl hydroxylase (PHD) 3 led to disruption of the intestinal epithelial barrier function. However, the underlying mechanism remains elusive. Here, we demonstrate that PHD3 controls the generation of intestine goblet cell. We found that genetic ablation of Phd3 in mice intestine epithelial cells reduced the amount of goblet cells. Mechanistically, PHD3 bounds the E3 ubiquitin ligase HUWE1 and prevented HUWE1 from mediating ubiquitination and degradation of ATOH1, an essential driver for goblet cell differentiation. The prolyl hydroxylase activity-deficient variant PHD3(H196A) also prevented ATOH1 destruction. A genetic intestine epithelial PHD3(H196A)-knockin had no effect on ATOH1 expression or goblet cell amount in mice, suggesting that the PHD3 prolyl hydroxylase activity is dispensable for its ability to control ATOH1 expression and goblet cell generation. In dextran sulfate sodium (DSS)-induced experimental colitis, PHD3-knockout rather than PHD3(H196A)-knockin sensitized the mice to DSS treatment. Our results reveal an additional critical mechanism underlying the regulation of ATOH1 expression and goblet cell generation and highlight that PHD3 plays a role in controlling intestine goblet cell generation in a hydroxylase-independent manner.
中文翻译:

脯氨酰羟化酶 3 通过稳定 ATOH1 来控制肠道杯状细胞的生成。
肠上皮不断自我更新并产生分化细胞,如秘书杯状细胞。肠道杯状细胞分泌形成凝胶的粘蛋白,形成粘液以形成防御屏障。我们之前报道过脯氨酰羟化酶 (PHD) 3 的缺失导致肠上皮屏障功能的破坏。然而,潜在的机制仍然难以捉摸。在这里,我们证明 PHD3 控制肠道杯状细胞的产生。我们发现小鼠肠上皮细胞中 Phd3 的基因消融减少了杯状细胞的数量。从机制上讲,PHD3 结合 E3 泛素连接酶 HUWE1 并阻止 HUWE1 介导 ATOH1 的泛素化和降解,ATOH1 是杯状细胞分化的重要驱动因素。脯氨酰羟化酶活性缺陷变体 PHD3(H196A) 也阻止了 ATOH1 的破坏。遗传性肠上皮 PHD3(H196A)-敲入蛋白对小鼠 ATOH1 表达或杯状细胞数量没有影响,表明 PHD3 脯氨酰羟化酶活性对于控制 ATOH1 表达和杯状细胞生成的能力是可有可无的。在葡聚糖硫酸钠 (DSS) 诱导的实验性结肠炎中,PHD3 敲除而不是 PHD3(H196A) 敲入使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。遗传性肠上皮 PHD3(H196A)-敲入蛋白对小鼠 ATOH1 表达或杯状细胞数量没有影响,表明 PHD3 脯氨酰羟化酶活性对于控制 ATOH1 表达和杯状细胞生成的能力是可有可无的。在葡聚糖硫酸钠 (DSS) 诱导的实验性结肠炎中,PHD3 敲除而不是 PHD3(H196A) 敲入使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。遗传性肠上皮 PHD3(H196A)-敲入蛋白对小鼠 ATOH1 表达或杯状细胞数量没有影响,表明 PHD3 脯氨酰羟化酶活性对于控制 ATOH1 表达和杯状细胞生成的能力是可有可无的。在葡聚糖硫酸钠 (DSS) 诱导的实验性结肠炎中,PHD3 敲除而不是 PHD3(H196A) 敲入使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。PHD3 基因敲除而不是 PHD3(H196A) 基因敲除使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。PHD3 基因敲除而不是 PHD3(H196A) 基因敲除使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。
更新日期:2020-01-20
中文翻译:
脯氨酰羟化酶 3 通过稳定 ATOH1 来控制肠道杯状细胞的生成。
肠上皮不断自我更新并产生分化细胞,如秘书杯状细胞。肠道杯状细胞分泌形成凝胶的粘蛋白,形成粘液以形成防御屏障。我们之前报道过脯氨酰羟化酶 (PHD) 3 的缺失导致肠上皮屏障功能的破坏。然而,潜在的机制仍然难以捉摸。在这里,我们证明 PHD3 控制肠道杯状细胞的产生。我们发现小鼠肠上皮细胞中 Phd3 的基因消融减少了杯状细胞的数量。从机制上讲,PHD3 结合 E3 泛素连接酶 HUWE1 并阻止 HUWE1 介导 ATOH1 的泛素化和降解,ATOH1 是杯状细胞分化的重要驱动因素。脯氨酰羟化酶活性缺陷变体 PHD3(H196A) 也阻止了 ATOH1 的破坏。遗传性肠上皮 PHD3(H196A)-敲入蛋白对小鼠 ATOH1 表达或杯状细胞数量没有影响,表明 PHD3 脯氨酰羟化酶活性对于控制 ATOH1 表达和杯状细胞生成的能力是可有可无的。在葡聚糖硫酸钠 (DSS) 诱导的实验性结肠炎中,PHD3 敲除而不是 PHD3(H196A) 敲入使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。遗传性肠上皮 PHD3(H196A)-敲入蛋白对小鼠 ATOH1 表达或杯状细胞数量没有影响,表明 PHD3 脯氨酰羟化酶活性对于控制 ATOH1 表达和杯状细胞生成的能力是可有可无的。在葡聚糖硫酸钠 (DSS) 诱导的实验性结肠炎中,PHD3 敲除而不是 PHD3(H196A) 敲入使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。遗传性肠上皮 PHD3(H196A)-敲入蛋白对小鼠 ATOH1 表达或杯状细胞数量没有影响,表明 PHD3 脯氨酰羟化酶活性对于控制 ATOH1 表达和杯状细胞生成的能力是可有可无的。在葡聚糖硫酸钠 (DSS) 诱导的实验性结肠炎中,PHD3 敲除而不是 PHD3(H196A) 敲入使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。PHD3 基因敲除而不是 PHD3(H196A) 基因敲除使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。PHD3 基因敲除而不是 PHD3(H196A) 基因敲除使小鼠对 DSS 治疗敏感。我们的研究结果揭示了调节 ATOH1 表达和杯状细胞生成的另一个关键机制,并强调 PHD3 以不依赖羟化酶的方式在控制肠道杯状细胞生成中发挥作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号