当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Identification of Highest-Affinity Binding Sites of Yeast Transcription Factor Families.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-16 , DOI: 10.1021/acs.jcim.9b01012 Zongyu Wang 1 , Wenying He 1 , Jijun Tang 1, 2, 3 , Fei Guo 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-16 , DOI: 10.1021/acs.jcim.9b01012 Zongyu Wang 1 , Wenying He 1 , Jijun Tang 1, 2, 3 , Fei Guo 1
Affiliation
|
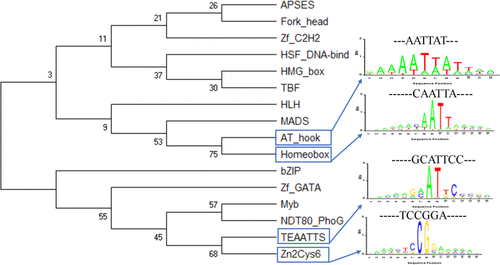
Transcription factors (TFs) play a crucial role in controlling key cellular processes and responding to the environment. Yeast is a single-cell fungal organism that is a vital biological model organism for studying transcription and translation in basic biology. The transcriptional control process of yeast cells has been extensively calculated and studied using traditional methods and high-throughput technologies. However, the identities of transcription factors that regulate major functional categories of genes remain unknown. Due to the avalanche of biological data in the post-genomic era, it is an urgent need to develop automated computational methods to enable accurate identification of efficient transcription factor binding sites from the large number of candidates. In this paper, we analyzed high-resolution DNA-binding profiles and motifs for TFs, covering all possible contiguous 8-mers. First, we divided all 8-mer motifs into 16 various categories and selected all sorts of samples from each category by setting the threshold of E-score. Then, we employed five feature representation methods. Also, we adopted a total of four feature selection methods to filter out useless features. Finally, we used Extreme Gradient Boosting (XGBoost) as our base classifier and then utilized the one-vs-rest tactics to build 16 binary classifiers to solve this multiclassification problem. In the experiment, our method achieved the best performance with an overall accuracy of 79.72% and Mathew's correlation coefficient of 0.77. We found the similarity relationship among each category from different TF families and obtained sequence motif schematic diagrams via multiple sequence alignment. The complexity of DNA recognition may act as an important role in the evolution of gene regulation. Source codes are available at https://github.com/guofei-tju/tfbs .
中文翻译:

酵母转录因子家族的最高亲和力结合位点的鉴定。
转录因子(TFs)在控制关键细胞过程和对环境的响应中起着至关重要的作用。酵母是一种单细胞真菌生物,是一种重要的生物学模型生物,用于研究基础生物学中的转录和翻译。酵母细胞的转录控制过程已使用传统方法和高通量技术进行了广泛的计算和研究。但是,调节基因的主要功能类别的转录因子的身份仍然未知。由于后基因组时代的大量生物数据,迫切需要开发自动化计算方法,以能够从大量候选物中准确识别有效的转录因子结合位点。在本文中,我们分析了TF的高分辨率DNA结合谱和基序,涵盖了所有可能的连续8聚体。首先,我们将所有8-mer主题划分为16个不同的类别,并通过设置E分数的阈值从每个类别中选择各种样本。然后,我们采用了五种特征表示方法。此外,我们总共采用了四种特征选择方法来过滤掉无用的特征。最后,我们使用极限梯度增强(XGBoost)作为我们的基本分类器,然后利用“一对多”策略构建16个二进制分类器来解决此多分类问题。在实验中,我们的方法以79.72%的整体准确度和0.77的马修相关系数获得了最佳性能。我们发现了来自不同TF家族的每个类别之间的相似关系,并通过多重序列比对获得了序列基序示意图。DNA识别的复杂性可能在基因调控的进化中起重要作用。源代码位于https://github.com/guofei-tju/tfbs。
更新日期:2020-01-16
中文翻译:
酵母转录因子家族的最高亲和力结合位点的鉴定。
转录因子(TFs)在控制关键细胞过程和对环境的响应中起着至关重要的作用。酵母是一种单细胞真菌生物,是一种重要的生物学模型生物,用于研究基础生物学中的转录和翻译。酵母细胞的转录控制过程已使用传统方法和高通量技术进行了广泛的计算和研究。但是,调节基因的主要功能类别的转录因子的身份仍然未知。由于后基因组时代的大量生物数据,迫切需要开发自动化计算方法,以能够从大量候选物中准确识别有效的转录因子结合位点。在本文中,我们分析了TF的高分辨率DNA结合谱和基序,涵盖了所有可能的连续8聚体。首先,我们将所有8-mer主题划分为16个不同的类别,并通过设置E分数的阈值从每个类别中选择各种样本。然后,我们采用了五种特征表示方法。此外,我们总共采用了四种特征选择方法来过滤掉无用的特征。最后,我们使用极限梯度增强(XGBoost)作为我们的基本分类器,然后利用“一对多”策略构建16个二进制分类器来解决此多分类问题。在实验中,我们的方法以79.72%的整体准确度和0.77的马修相关系数获得了最佳性能。我们发现了来自不同TF家族的每个类别之间的相似关系,并通过多重序列比对获得了序列基序示意图。DNA识别的复杂性可能在基因调控的进化中起重要作用。源代码位于https://github.com/guofei-tju/tfbs。










































 京公网安备 11010802027423号
京公网安备 11010802027423号