当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Reaction Mechanism and Free Energy Barriers for the Chemisorption of CO2 by Ionic Entities.
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-01-23 , DOI: 10.1021/acs.jpca.9b06817 Koteswara Rao Gorantla 1 , Bhabani S Mallik 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-01-23 , DOI: 10.1021/acs.jpca.9b06817 Koteswara Rao Gorantla 1 , Bhabani S Mallik 1
Affiliation
|
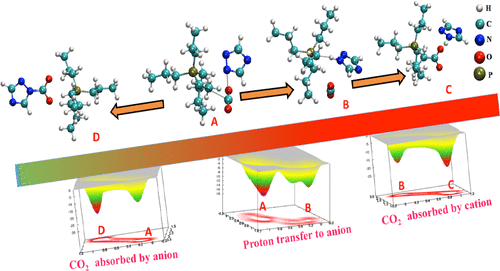
Ionic liquids, a class of alternative solvents, are known for their ability to capture carbon dioxide (CO2). The understanding of the role of the individual ionic entity of the ionic liquid (IL) and the involved mechanism is essential to design a better solvent for the capture process. In the present study, we employed density functional theory based electronic structure calculations and metadynamics method based first-principles molecular dynamics (FPMD) simulations to investigate the roles of the cation and anion of the IL by analyzing the energetics and free energy profile of the involved chemical reactions. The mechanism of chemisorption of CO2 by the aprotic N-heterocyclic and phenolate anions paired with tetrapropyl phosphonium cation [P3333] were studied to understand the reaction mechanism of the initial capture process. The process of uptaking of CO2 by the [P3333][1,2,4-Triz] was studied by the first-principles calculations. The transition states in the reaction pathways were computed by the synchronous transit-guided quasi-Newton method and confirmed by the intrinsic reaction coordinate calculations using first-principles simulations. The dynamics of the energetics of the chemisorption process were studied by constructing the free energy surface using metadynamics-based FPMD simulations. First, the nucleophilic center was generated at the α-carbon of the cation by transferring a proton to the anion with the formation of the phosphorus ylide. The formed cation ylide chemisorbs CO2 through the formation of a bond between the α-carbon of ylide and the carbon of CO2. The direct addition of CO2 to the anion of the ionic pair was studied as the second pathway. We find that the chemisorption of CO2 by the anion is more favorable than that by the cation. By comparing the chemisorption of CO2 by the ions, we observe that the deprotonation of the alkyl chain is the more deciding factor, which depends on the basicity of anion and the length of the alkyl chain. We computed the free energy landscapes for the ionic pairs by varying another four anions like cyclohexanolate, 2,4,6-trifluorophenolate, imidazolate, and benzotriazolide paired with tetrapropyl phosphonium cation. The effect of the alkyl chain on the proton transfer was studied by tetrabutyl and tetrapentyl phosphonium cations paired with 1,2,4-triazolide anion. The carbonated product, formed from the anion, is thermodynamically controlled, while the carboxylated product (formed from cation) is kinetically controlled. We hope that our findings will enhance the knowledge of the selectivity of ionic entities for designing IL-based solvents for the capture process of CO2.
中文翻译:

离子实体化学吸附CO2的反应机理和自由能壁垒。
离子液体是一类替代溶剂,因其捕获二氧化碳(CO2)的能力而闻名。了解离子液体(IL)的各个离子实体的作用及其所涉及的机理,对于为捕获过程设计更好的溶剂至关重要。在本研究中,我们通过基于密度泛函理论的电子结构计算和基于元动力学方法的第一性原理分子动力学(FPMD)模拟,通过分析所涉及分子的能量和自由能曲线来研究IL的阳离子和阴离子的作用。化学反应。研究了质子惰性的N-杂环和酚盐阴离子与四丙基propyl阳离子[P3333]配对对CO2的化学吸附机理,以了解初始捕获过程的反应机理。通过第一性原理计算研究了[P3333] [1,2,4-Triz]吸收二氧化碳的过程。反应路径中的过渡态通过同步过渡引导的拟牛顿法计算,并通过使用第一性原理模拟的内在反应坐标计算得到证实。通过使用基于元动力学的FPMD模拟构造自由能表面,研究了化学吸附过程的能量学动力学。首先,通过将质子转移到阴离子上并形成磷内鎓盐,在阳离子的α-碳处生成亲核中心。形成的阳离子内鎓盐通过内鎓盐的α-碳与CO2碳之间的键形成化学吸附CO2。研究了将CO2直接加到离子对阴离子中作为第二种途径。我们发现,阴离子对CO2的化学吸附比阳离子对化学吸附更有利。通过比较离子对CO 2的化学吸附,我们观察到烷基链的去质子化是更决定性的因素,这取决于阴离子的碱性和烷基链的长度。我们通过改变另外四个阴离子(如环己酸根,2,4,6-三氟酚酸根,咪唑酸根和苯并三偶氮根与四丙基phospho阳离子配对)来计算离子对的自由能态。通过四丁基和四戊基phospho阳离子与1,2,4-三唑化物阴离子配对,研究了烷基链对质子转移的影响。对由阴离子形成的碳酸化产物进行热力学控制,而对羧基化产物(由阳离子形成)进行动力学控制。
更新日期:2020-01-23
中文翻译:
离子实体化学吸附CO2的反应机理和自由能壁垒。
离子液体是一类替代溶剂,因其捕获二氧化碳(CO2)的能力而闻名。了解离子液体(IL)的各个离子实体的作用及其所涉及的机理,对于为捕获过程设计更好的溶剂至关重要。在本研究中,我们通过基于密度泛函理论的电子结构计算和基于元动力学方法的第一性原理分子动力学(FPMD)模拟,通过分析所涉及分子的能量和自由能曲线来研究IL的阳离子和阴离子的作用。化学反应。研究了质子惰性的N-杂环和酚盐阴离子与四丙基propyl阳离子[P3333]配对对CO2的化学吸附机理,以了解初始捕获过程的反应机理。通过第一性原理计算研究了[P3333] [1,2,4-Triz]吸收二氧化碳的过程。反应路径中的过渡态通过同步过渡引导的拟牛顿法计算,并通过使用第一性原理模拟的内在反应坐标计算得到证实。通过使用基于元动力学的FPMD模拟构造自由能表面,研究了化学吸附过程的能量学动力学。首先,通过将质子转移到阴离子上并形成磷内鎓盐,在阳离子的α-碳处生成亲核中心。形成的阳离子内鎓盐通过内鎓盐的α-碳与CO2碳之间的键形成化学吸附CO2。研究了将CO2直接加到离子对阴离子中作为第二种途径。我们发现,阴离子对CO2的化学吸附比阳离子对化学吸附更有利。通过比较离子对CO 2的化学吸附,我们观察到烷基链的去质子化是更决定性的因素,这取决于阴离子的碱性和烷基链的长度。我们通过改变另外四个阴离子(如环己酸根,2,4,6-三氟酚酸根,咪唑酸根和苯并三偶氮根与四丙基phospho阳离子配对)来计算离子对的自由能态。通过四丁基和四戊基phospho阳离子与1,2,4-三唑化物阴离子配对,研究了烷基链对质子转移的影响。对由阴离子形成的碳酸化产物进行热力学控制,而对羧基化产物(由阳离子形成)进行动力学控制。










































 京公网安备 11010802027423号
京公网安备 11010802027423号