当前位置:
X-MOL 学术
›
PLOS Pathog.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Quantifying within-host diversity of H5N1 influenza viruses in humans and poultry in Cambodia.
PLoS Pathogens ( IF 5.5 ) Pub Date : 2020-01-17 , DOI: 10.1371/journal.ppat.1008191 Louise H Moncla 1 , Trevor Bedford 1, 2 , Philippe Dussart 3 , Srey Viseth Horm 3 , Sareth Rith 3 , Philippe Buchy 4 , Erik A Karlsson 3 , Lifeng Li 5, 6 , Yongmei Liu 5, 6 , Huachen Zhu 5, 6 , Yi Guan 5, 6 , Thomas C Friedrich 7, 8 , Paul F Horwood 3, 9
PLoS Pathogens ( IF 5.5 ) Pub Date : 2020-01-17 , DOI: 10.1371/journal.ppat.1008191 Louise H Moncla 1 , Trevor Bedford 1, 2 , Philippe Dussart 3 , Srey Viseth Horm 3 , Sareth Rith 3 , Philippe Buchy 4 , Erik A Karlsson 3 , Lifeng Li 5, 6 , Yongmei Liu 5, 6 , Huachen Zhu 5, 6 , Yi Guan 5, 6 , Thomas C Friedrich 7, 8 , Paul F Horwood 3, 9
Affiliation
|
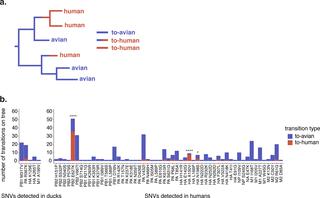
Avian influenza viruses (AIVs) periodically cross species barriers and infect humans. The likelihood that an AIV will evolve mammalian transmissibility depends on acquiring and selecting mutations during spillover, but data from natural infection is limited. We analyze deep sequencing data from infected humans and domestic ducks in Cambodia to examine how H5N1 viruses evolve during spillover. Overall, viral populations in both species are predominated by low-frequency (<10%) variation shaped by purifying selection and genetic drift, and half of the variants detected within-host are never detected on the H5N1 virus phylogeny. However, we do detect a subset of mutations linked to human receptor binding and replication (PB2 E627K, HA A150V, and HA Q238L) that arose in multiple, independent humans. PB2 E627K and HA A150V were also enriched along phylogenetic branches leading to human infections, suggesting that they are likely human-adaptive. Our data show that H5N1 viruses generate putative human-adapting mutations during natural spillover infection, many of which are detected at >5% frequency within-host. However, short infection times, genetic drift, and purifying selection likely restrict their ability to evolve extensively during a single infection. Applying evolutionary methods to sequence data, we reveal a detailed view of H5N1 virus adaptive potential, and develop a foundation for studying host-adaptation in other zoonotic viruses.
中文翻译:

在柬埔寨量化人类和家禽中H5N1流感病毒的宿主内部多样性。
禽流感病毒(AIV)会定期越过物种壁垒并感染人类。AIV进化出哺乳动物可传播性的可能性取决于溢出过程中获得和选择突变,但是来自自然感染的数据是有限的。我们分析了来自柬埔寨受感染人类和家养鸭子的深度测序数据,以检查H5N1病毒在溢出过程中如何进化。总体而言,两个物种中的病毒种群均以通过纯化选择和遗传漂移而形成的低频变异(<10%)为主,而且在H5N1病毒系统发育中从未检测到宿主内部检测到的一半变异。但是,我们确实检测到与人类受体结合和复制有关的突变子集(PB2 E627K,HA A150V和HA Q238L),这些突变发生于多个独立的人类中。PB2 E627K和HA A150V也沿导致人类感染的系统发育分支富集,表明它们很可能是人类适应性的。我们的数据表明,H5N1病毒在自然溢出感染期间会产生推测的人类适应性突变,其中许多在宿主体内的检出频率高于5%。但是,短的感染时间,遗传漂移和纯化选择可能会限制它们在单次感染中广泛进化的能力。将进化方法应用于序列数据,我们揭示了H5N1病毒的适应潜力的详细视图,并为研究其他人畜共患病毒的宿主适应性奠定了基础。我们的数据表明,H5N1病毒在自然溢出感染期间会产生推测的人类适应性突变,其中许多在宿主体内的检出频率高于5%。但是,短的感染时间,遗传漂移和纯化选择可能会限制它们在单次感染中广泛进化的能力。将进化方法应用于序列数据,我们揭示了H5N1病毒的适应潜力的详细视图,并为研究其他人畜共患病毒的宿主适应性奠定了基础。我们的数据表明,H5N1病毒在自然溢出感染期间会产生推测的人类适应性突变,其中许多在宿主体内的检出频率高于5%。但是,短的感染时间,遗传漂移和纯化选择可能会限制它们在单次感染期间广泛进化的能力。将进化方法应用于序列数据,我们揭示了H5N1病毒的适应潜力的详细视图,并为研究其他人畜共患病毒的宿主适应性奠定了基础。
更新日期:2020-01-17
中文翻译:
在柬埔寨量化人类和家禽中H5N1流感病毒的宿主内部多样性。
禽流感病毒(AIV)会定期越过物种壁垒并感染人类。AIV进化出哺乳动物可传播性的可能性取决于溢出过程中获得和选择突变,但是来自自然感染的数据是有限的。我们分析了来自柬埔寨受感染人类和家养鸭子的深度测序数据,以检查H5N1病毒在溢出过程中如何进化。总体而言,两个物种中的病毒种群均以通过纯化选择和遗传漂移而形成的低频变异(<10%)为主,而且在H5N1病毒系统发育中从未检测到宿主内部检测到的一半变异。但是,我们确实检测到与人类受体结合和复制有关的突变子集(PB2 E627K,HA A150V和HA Q238L),这些突变发生于多个独立的人类中。PB2 E627K和HA A150V也沿导致人类感染的系统发育分支富集,表明它们很可能是人类适应性的。我们的数据表明,H5N1病毒在自然溢出感染期间会产生推测的人类适应性突变,其中许多在宿主体内的检出频率高于5%。但是,短的感染时间,遗传漂移和纯化选择可能会限制它们在单次感染中广泛进化的能力。将进化方法应用于序列数据,我们揭示了H5N1病毒的适应潜力的详细视图,并为研究其他人畜共患病毒的宿主适应性奠定了基础。我们的数据表明,H5N1病毒在自然溢出感染期间会产生推测的人类适应性突变,其中许多在宿主体内的检出频率高于5%。但是,短的感染时间,遗传漂移和纯化选择可能会限制它们在单次感染中广泛进化的能力。将进化方法应用于序列数据,我们揭示了H5N1病毒的适应潜力的详细视图,并为研究其他人畜共患病毒的宿主适应性奠定了基础。我们的数据表明,H5N1病毒在自然溢出感染期间会产生推测的人类适应性突变,其中许多在宿主体内的检出频率高于5%。但是,短的感染时间,遗传漂移和纯化选择可能会限制它们在单次感染期间广泛进化的能力。将进化方法应用于序列数据,我们揭示了H5N1病毒的适应潜力的详细视图,并为研究其他人畜共患病毒的宿主适应性奠定了基础。










































 京公网安备 11010802027423号
京公网安备 11010802027423号