当前位置:
X-MOL 学术
›
npj Genom. Med.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The impact of adjusting for baseline in pharmacogenomic genome-wide association studies of quantitative change.
npj Genomic Medicine ( IF 4.7 ) Pub Date : 2020-01-16 , DOI: 10.1038/s41525-019-0109-4 Akinyemi Oni-Orisan 1, 2 , Tanushree Haldar 2 , Dilrini K Ranatunga 3 , Marisa W Medina 4 , Catherine Schaefer 3 , Ronald M Krauss 4, 5 , Carlos Iribarren 3 , Neil Risch 2, 3, 6 , Thomas J Hoffmann 2, 6
npj Genomic Medicine ( IF 4.7 ) Pub Date : 2020-01-16 , DOI: 10.1038/s41525-019-0109-4 Akinyemi Oni-Orisan 1, 2 , Tanushree Haldar 2 , Dilrini K Ranatunga 3 , Marisa W Medina 4 , Catherine Schaefer 3 , Ronald M Krauss 4, 5 , Carlos Iribarren 3 , Neil Risch 2, 3, 6 , Thomas J Hoffmann 2, 6
Affiliation
|
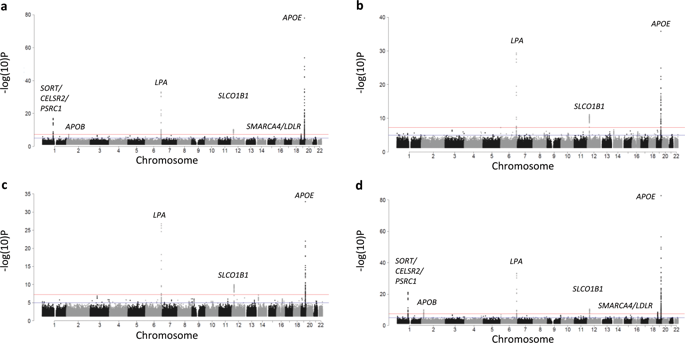
In pharmacogenomic studies of quantitative change, any association between genetic variants and the pretreatment (baseline) measurement can bias the estimate of effect between those variants and drug response. A putative solution is to adjust for baseline. We conducted a series of genome-wide association studies (GWASs) for low-density lipoprotein cholesterol (LDL-C) response to statin therapy in 34,874 participants of the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort as a case study to investigate the impact of baseline adjustment on results generated from pharmacogenomic studies of quantitative change. Across phenotypes of statin-induced LDL-C change, baseline adjustment identified variants from six loci meeting genome-wide significance (SORT/CELSR2/PSRC1, LPA, SLCO1B1, APOE, APOB, and SMARCA4/LDLR). In contrast, baseline-unadjusted analyses yielded variants from three loci meeting the criteria for genome-wide significance (LPA, APOE, and SLCO1B1). A genome-wide heterogeneity test of baseline versus statin on-treatment LDL-C levels was performed as the definitive test for the true effect of genetic variants on statin-induced LDL-C change. These findings were generally consistent with the models not adjusting for baseline signifying that genome-wide significant hits generated only from baseline-adjusted analyses (SORT/CELSR2/PSRC1, APOB, SMARCA4/LDLR) were likely biased. We then comprehensively reviewed published GWASs of drug-induced quantitative change and discovered that more than half (59%) inappropriately adjusted for baseline. Altogether, we demonstrate that (1) baseline adjustment introduces bias in pharmacogenomic studies of quantitative change and (2) this erroneous methodology is highly prevalent. We conclude that it is critical to avoid this common statistical approach in future pharmacogenomic studies of quantitative change.
中文翻译:

调整药物基因组全基因组关联研究中基线的影响。
在数量变化的药物基因组学研究中,遗传变异与预处理(基线)之间的任何关联都可能会使这些变异与药物反应之间的效应估计产生偏差。一个公认的解决方案是调整基线。我们针对34,874名成人健康与衰老遗传流行病学研究(GERA)队列中的参与者,对低密度脂蛋白胆固醇(LDL-C)对他汀类药物治疗的反应进行了全基因组关联研究(GWAS),研究基线调整对药物基因组学定量变化研究结果的影响。在他汀类药物诱导的LDL-C变化的各种表型中,基线调整从六个基因组中鉴定出满足全基因组意义的变体(SORT / CELSR2 / PSRC1,LPA,SLCO1B1,APOE,APOB和SMARCA4 / LDLR)。相反,基线未经调整的分析产生了来自三个基因座的变体,这些变体符合全基因组重要性的标准(LPA,APOE和SLCO1B1)。进行了基线水平与他汀治疗后LDL-C水平的全基因组异质性测试,作为遗传变异对他汀诱导的LDL-C变化的真实影响的确定性测试。这些发现通常与未针对基线进行调整的模型一致,这表明仅由基线调整后的分析(SORT / CELSR2 / PSRC1,APOB,SMARCA4 / LDLR)产生的全基因组范围内的重大命中可能存在偏差。然后,我们全面审查了已发表的有关药物引起的定量变化的GWAS,发现超过一半(59%)的基线调整不当。共,我们证明(1)基线调整在药物基因组学研究中对定量变化产生了偏见,(2)这种错误的方法非常普遍。我们得出结论,在以后的定量变化药物基因组学研究中避免使用这种通用的统计方法至关重要。
更新日期:2020-01-16
中文翻译:
调整药物基因组全基因组关联研究中基线的影响。
在数量变化的药物基因组学研究中,遗传变异与预处理(基线)之间的任何关联都可能会使这些变异与药物反应之间的效应估计产生偏差。一个公认的解决方案是调整基线。我们针对34,874名成人健康与衰老遗传流行病学研究(GERA)队列中的参与者,对低密度脂蛋白胆固醇(LDL-C)对他汀类药物治疗的反应进行了全基因组关联研究(GWAS),研究基线调整对药物基因组学定量变化研究结果的影响。在他汀类药物诱导的LDL-C变化的各种表型中,基线调整从六个基因组中鉴定出满足全基因组意义的变体(SORT / CELSR2 / PSRC1,LPA,SLCO1B1,APOE,APOB和SMARCA4 / LDLR)。相反,基线未经调整的分析产生了来自三个基因座的变体,这些变体符合全基因组重要性的标准(LPA,APOE和SLCO1B1)。进行了基线水平与他汀治疗后LDL-C水平的全基因组异质性测试,作为遗传变异对他汀诱导的LDL-C变化的真实影响的确定性测试。这些发现通常与未针对基线进行调整的模型一致,这表明仅由基线调整后的分析(SORT / CELSR2 / PSRC1,APOB,SMARCA4 / LDLR)产生的全基因组范围内的重大命中可能存在偏差。然后,我们全面审查了已发表的有关药物引起的定量变化的GWAS,发现超过一半(59%)的基线调整不当。共,我们证明(1)基线调整在药物基因组学研究中对定量变化产生了偏见,(2)这种错误的方法非常普遍。我们得出结论,在以后的定量变化药物基因组学研究中避免使用这种通用的统计方法至关重要。










































 京公网安备 11010802027423号
京公网安备 11010802027423号