当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Gain-Scanning for Protein Microarray Assays.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-01-13 , DOI: 10.1021/acs.jproteome.9b00892 Feng Feng 1 , Sila Toksoz Ataca 1 , Mingxuan Ran 1 , Yumei Wang 1 , Michael Breen 1 , Thomas B Kepler 1, 2
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-01-13 , DOI: 10.1021/acs.jproteome.9b00892 Feng Feng 1 , Sila Toksoz Ataca 1 , Mingxuan Ran 1 , Yumei Wang 1 , Michael Breen 1 , Thomas B Kepler 1, 2
Affiliation
|
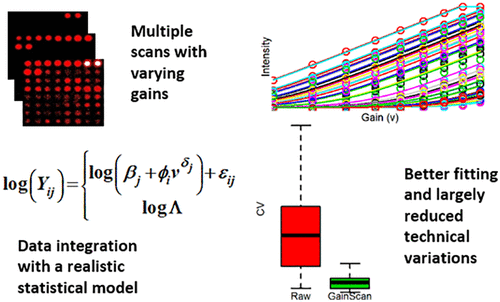
Protein microarrays consist of known proteins spotted onto solid substrates and are used to perform highly multivariate assessments of protein-binding interactions. Human protein arrays are routinely applied to pathogen detection, immune response biomarker profiling, and antibody specificity profiling. Here, we describe and demonstrate a new data processing procedure, gain-scan, in which data were acquired under multiple photomultiplier tube (PMT) settings, followed by data fitting with a power function model to estimate the incident light signals of the array spots. Data acquisition under multiple PMT settings solves the difficulty of determining the single optimal PMT gain setting and allows us to maximize the detection of low-intensity signals while avoiding the saturation of high-intensity ones at the same time. The gain-scan data acquisition and fitting also significantly lower the variances over the detectable range of signals and improve the linear data normalization. The performance of the proposed procedure was verified by analyzing the profiling data of both the human polyclonal serum samples and the monoclonal antibody samples with both technical replicates and biological replicates. We showed that the multigain power function was an appropriate model for describing data acquired under multiple PMT settings. The gain-scan fitting alone or in combination with the linear normalization could effectively reduce the technical variability of the array data and lead to better sample separability and more sensitive differential analysis.
中文翻译:

蛋白质芯片分析的增益扫描。
蛋白质微阵列由点样在固体基质上的已知蛋白质组成,用于对蛋白质结合相互作用进行高度多变量评估。人类蛋白质阵列通常应用于病原体检测,免疫反应生物标志物分析和抗体特异性分析。在这里,我们描述并演示了一种新的数据处理程序,即增益扫描,其中在多个光电倍增管(PMT)设置下获取数据,然后对数据进行幂函数模型拟合以估计阵列点的入射光信号。在多个PMT设置下进行数据采集解决了确定单个最佳PMT增益设置的困难,并使我们能够最大程度地检测低强度信号,同时避免高强度信号的饱和。增益扫描数据采集和拟合还可以显着降低信号可检测范围内的方差,并改善线性数据归一化。通过分析人多克隆血清样品和单克隆抗体样品(具有技术重复和生物学重复)的分析数据,验证了所提出程序的性能。我们表明,多增益幂函数是用于描述在多个PMT设置下获取的数据的合适模型。单独或与线性归一化结合使用增益扫描拟合可以有效地减少阵列数据的技术变异性,并带来更好的样品可分离性和更灵敏的差异分析。
更新日期:2020-01-13
中文翻译:
蛋白质芯片分析的增益扫描。
蛋白质微阵列由点样在固体基质上的已知蛋白质组成,用于对蛋白质结合相互作用进行高度多变量评估。人类蛋白质阵列通常应用于病原体检测,免疫反应生物标志物分析和抗体特异性分析。在这里,我们描述并演示了一种新的数据处理程序,即增益扫描,其中在多个光电倍增管(PMT)设置下获取数据,然后对数据进行幂函数模型拟合以估计阵列点的入射光信号。在多个PMT设置下进行数据采集解决了确定单个最佳PMT增益设置的困难,并使我们能够最大程度地检测低强度信号,同时避免高强度信号的饱和。增益扫描数据采集和拟合还可以显着降低信号可检测范围内的方差,并改善线性数据归一化。通过分析人多克隆血清样品和单克隆抗体样品(具有技术重复和生物学重复)的分析数据,验证了所提出程序的性能。我们表明,多增益幂函数是用于描述在多个PMT设置下获取的数据的合适模型。单独或与线性归一化结合使用增益扫描拟合可以有效地减少阵列数据的技术变异性,并带来更好的样品可分离性和更灵敏的差异分析。










































 京公网安备 11010802027423号
京公网安备 11010802027423号