当前位置:
X-MOL 学术
›
J. Biomed. Inform.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Compositional framework for multitask learning in the identification of cleavage sites of HIV-1 protease.
Journal of Biomedical informatics ( IF 4.0 ) Pub Date : 2020-01-11 , DOI: 10.1016/j.jbi.2020.103376 Deepak Singh 1 , Dilip Singh Sisodia 1 , Pradeep Singh 1
Journal of Biomedical informatics ( IF 4.0 ) Pub Date : 2020-01-11 , DOI: 10.1016/j.jbi.2020.103376 Deepak Singh 1 , Dilip Singh Sisodia 1 , Pradeep Singh 1
Affiliation
|
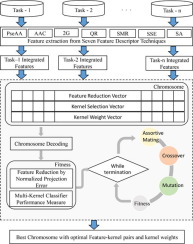
Inadequate patient samples and costly annotated data generations result into the smaller dataset in the biomedical domain. Due to which the predictions with a trained model that usually reveal a single small dataset association are fail to derive robust insights. To cope with the data sparsity, a promising strategy of combining data from the different related tasks is exercised in various application. Motivated by, successful work in the various bioinformatics application, we propose a multitask learning model based on multi-kernel that exploits the dependencies among various related tasks. This work aims to combine the knowledge from experimental studies of the different dataset to build stronger predictive models for HIV-1 protease cleavage sites prediction. In this study, a set of peptide data from one source is referred as 'task' and to integrate interactions from multiple tasks; our method exploits the common features and parameters sharing across the data source. The proposed framework uses feature integration, feature selection, multi-kernel and multifactorial evolutionary algorithm to model multitask learning. The framework considered seven different feature descriptors and four different kernel variants of support vector machines to form the optimal multi-kernel learning model. To validate the effectiveness of the model, the performance parameters such as average accuracy, and area under curve have been evaluated on the suggested model. We also carried out Friedman and post hoc statistical test to substantiate the significant improvement achieved by the proposed framework. The result obtained following the extensive experiment confirms the belief that multitask learning in cleavage site identification can improve the performance.
中文翻译:

用于识别 HIV-1 蛋白酶切割位点的多任务学习的组成框架。
不充分的患者样本和昂贵的注释数据生成导致生物医学领域的数据集较小。因此,使用通常显示单个小数据集关联的训练模型的预测无法获得可靠的见解。为了应对数据稀疏性,在各种应用中采用了一种有前途的策略,即组合来自不同相关任务的数据。受到各种生物信息学应用的成功工作的启发,我们提出了一种基于多内核的多任务学习模型,该模型利用了各种相关任务之间的依赖关系。这项工作旨在结合来自不同数据集的实验研究的知识,为 HIV-1 蛋白酶切割位点预测建立更强大的预测模型。在这项研究中,来自一个来源的一组肽数据被称为“任务” 并整合来自多个任务的交互;我们的方法利用了跨数据源共享的共同特征和参数。所提出的框架使用特征集成、特征选择、多内核和多因素进化算法来模拟多任务学习。该框架考虑了七种不同的特征描述符和四种不同的支持向量机内核变体,以形成最佳的多内核学习模型。为了验证模型的有效性,对建议模型的平均精度和曲线下面积等性能参数进行了评估。我们还进行了弗里德曼和事后统计检验,以证实所提出的框架取得的显着改进。
更新日期:2020-01-13
中文翻译:
用于识别 HIV-1 蛋白酶切割位点的多任务学习的组成框架。
不充分的患者样本和昂贵的注释数据生成导致生物医学领域的数据集较小。因此,使用通常显示单个小数据集关联的训练模型的预测无法获得可靠的见解。为了应对数据稀疏性,在各种应用中采用了一种有前途的策略,即组合来自不同相关任务的数据。受到各种生物信息学应用的成功工作的启发,我们提出了一种基于多内核的多任务学习模型,该模型利用了各种相关任务之间的依赖关系。这项工作旨在结合来自不同数据集的实验研究的知识,为 HIV-1 蛋白酶切割位点预测建立更强大的预测模型。在这项研究中,来自一个来源的一组肽数据被称为“任务” 并整合来自多个任务的交互;我们的方法利用了跨数据源共享的共同特征和参数。所提出的框架使用特征集成、特征选择、多内核和多因素进化算法来模拟多任务学习。该框架考虑了七种不同的特征描述符和四种不同的支持向量机内核变体,以形成最佳的多内核学习模型。为了验证模型的有效性,对建议模型的平均精度和曲线下面积等性能参数进行了评估。我们还进行了弗里德曼和事后统计检验,以证实所提出的框架取得的显着改进。










































 京公网安备 11010802027423号
京公网安备 11010802027423号