当前位置:
X-MOL 学术
›
Process Biochem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Combined metabolomic and transcriptomic analysis reveals key candidate genes involved in the regulation of flavonoid accumulation in Anoectochilus roxburghii
Process Biochemistry ( IF 4.4 ) Pub Date : 2020-04-01 , DOI: 10.1016/j.procbio.2020.01.004 Ying Chen , Wangyun Pan , Sha Jin , Sizu Lin
Process Biochemistry ( IF 4.4 ) Pub Date : 2020-04-01 , DOI: 10.1016/j.procbio.2020.01.004 Ying Chen , Wangyun Pan , Sha Jin , Sizu Lin
|
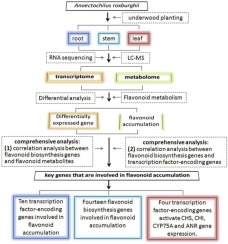
Abstract Anoectochilus roxburghii, a species used in Chinese herbal medicine, has unique characteristics. This plant has important medicinal and ornamental value and is distributed primarily in China, including Fujian, Zhejiang, Jiangxi, and Guizhou provinces, and in Taiwan. Flavonoids are involved in leaf pigment formation and are major pharmacodynamic substances. However, the molecular mechanisms that regulate accumulation of flavonoids remain unclear, which has significantly limited their application. To elucidate the molecular mechanisms underlying the regulation of flavonoid accumulation in A. roxburghii, root, stem and leaf samples were collected for constructing transcriptomic and metabolomic datasets using RNA sequencing and liquid chromatography-mass spectrometry (LC–MS) techniques. Sequencing of the transcriptomes of the different organs generated 60.2 Gb of data, which were assembled into 186,865 unigenes. Metabolomic analysis resulted in the identification of 10,690 metabolites. Based on the ESI+ mode, 4,010, 4,008 and 4,013 metabolites were annotated in roots, stems, and leaves, respectively; based on the ESI– mode, 1,530, 1,530, and 1,531 metabolites were annotated, respectively. Differential analyses of the transcriptome and flavonoid metabolism of the different organs revealed the greatest significant differences between roots and leaves, followed by differences between stems and leaves; differences between roots and stems were the smallest. According to analysis of differentially expressed genes (DEGs), the secondary metabolism-related DEGs between roots and stems, between roots and leaves, and between leaves and stems were classified into 16, 21, and 19 secondary metabolic Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, respectively. Among these pathways, flavonoid biosynthesis, flavone and flavonol biosynthesis, and isoflavonoid biosynthesis significantly differed in terms of both gene expression and secondary metabolism. Moreover, the key genes involved in flavonoid accumulation were comprehensively analyzed using metabolic and transcriptomic data, and ten transcription factor-encoding genes and fourteen flavonoid biosynthesis genes were identified. Specifically, four of the ten transcription factor-encoding genes appear to activate CHS, CHI, CYP75A and ANR gene expression. The combined approach used in this study provides an in-depth understanding of the molecular regulatory mechanisms underlying flavonoid accumulation in A. roxburghii and is a powerful tool that can uncover valuable information for plant breeders.
更新日期:2020-04-01


























 京公网安备 11010802027423号
京公网安备 11010802027423号